
Abstract
Intraductal papillary neoplasm of the bile duct (IPNB) is a mass-forming neoplasm in the bile duct considered to be the biliary counterpart of pancreatic intraductal papillary mucinous neoplasm (IPMN). By its cell lineage, IPNB can be classified into gastric, intestinal, pancreatobiliary, and oncocytic types. Recently, a group of Japanese and Korean pathologists proposed that IPNB be classified into two types, with type 1, being the histological counterpart of IPMN and type 2, having a more complex histological architecture. We used targeted next-generation sequencing to study the molecular change of 37 IPNBs and identified frequent mutations of KRAS (49%), GNAS (32%), RNF43 (24%), APC (24%), TP53 (24%), and CTNNB1 (11%) in IPNBs. Intestinal-type IPNB was associated with KRAS, GNAS, and RNF43 mutations. Japan–Korea consensus type 1 was associated with KRAS and GNAS mutations. All four IPNBs with CTNNB1 mutations were of pancreatobiliary type and located in the extrahepatic bile duct. A hierarchical analysis identified three distinct groups within IPNB: group 1 was Japan–Korea consensus type 1 tumors with macroscopic mucin, old age, and frequent KRAS, GNAS, and RNF43 mutations. Group 2 was Japan–Korea consensus type 2 with intestinal differentiation and frequent KRAS mutation but rare GNAS mutation, MUC2 expression, and macroscopic mucin. Group 3 was characterized by CTNNB1 mutation, extrahepatic location, lack of expression of intestinal markers, Japan–Korea consensus type 2, and lack of mutations in KRAS, APC, RNF43, and GNAS. Our results indicated that IPNB is a heterogeneous disease and that the activation of Ras-mitogen-activated protein kinase (MAPK), Wnt/β-catenin, and G-protein-coupled receptor (GPCR)–cAMP signaling is the main oncogenic mechanism of IPNB.
Similar content being viewed by others

Introduction
Intraductal papillary neoplasm of the bile duct (IPNB) is a neoplastic lesion characterized by noninvasive papillary or villous biliary epithelial cells covering the fibrovascular stalk in dilated bile ducts. It is considered the biliary counterpart of the intraductal papillary mucinous neoplasm (IPMN) of the pancreas [1], and a precursor lesion of cholangiocarcinoma [2]. Unlike IPMN, IPNB is mainly reported in East Asian countries, such as Taiwan, Japan, and Korea [3,4,5,6]. IPNB is a heterogeneous disease. Reports have suggested the use of the classification system of IPMN to categorize IPNB into four cell lineage subtypes: gastric, intestinal, pancreatobiliary, and oncocytic [5]. Recently, a group of Japanese and Korean pathologists proposed a consensus classification of IPNB into two types [6]. Type 1 is a histological counterpart of IPMN and mainly located in the intrahepatic bile duct. Type 2 has a more complex histological architecture with irregular papillary branching or with foci of solid-tubular components and mainly involves the extrahepatic bile ducts [6]. Type 2 IPNB frequently exhibits high-grade dysplasia, and is more likely to have invasive adenocarcinoma at the time of surgical resection [6].
The molecular pathogenesis of IPNB remains to be clarified. We, along with other groups, have identified frequent KRAS, GNAS, and RNF43 mutations in IPNB, mainly in intestinal-type IPNB with mucin hypersecretion [4, 7,8,9]. For a more comprehensive analysis of the molecular pathogenesis of IPNB, we used a targeted next-generation sequencing panel which included commonly mutated genes in gastrointestinal adenocarcinoma. The objective was to identify novel mutated genes in IPNB and correlated the findings to the histological and clinical features.
Materials and methods
Patients
Thirty-seven patients with IPNB diagnosed from 1998 to 2015, and who underwent surgical resection in the National Taiwan University Hospital were enrolled in this study. The sections were reviewed by two hepatobiliary pathologists (J.-H.T. and Y.-M.J.) and classified according to cell lineage and to the Japan–Korea consensus classification [6]. Clinical data, including demographics, clinical information, and patient outcome, were obtained from chart review. This study was approved under the regulations of the Research Ethics Committee of the National Taiwan University Hospital (Approval number 201704003RIND), and was conducted according to the principles of the Declaration of Helsinki.
Next-generation sequencing
Genomic DNA was extracted from the tumorous areas macrodissected from 10-mm paraffin sections with a QIAamp® DNA FFPE Tissue Kit (QIAGEN, Santa Clarita, CA, USA) according to manufacturer protocol. A multiplex PCR target-enrichment panel for target relevant genes was used to enrich DNA by a custom-made QIAseq Targeted DNA Panel (QIAGEN). The genes and exons analyzed are listed in Supplementary Table 1. The primers used for DNA amplification were designed by QIAGEN QIAseq designer website (https://www.qiagen.com/us/shop/genes-and-pathways/custom-products/custom-array-products/qiaseq-targeted-dna-panels/#design). Specimens were macrodissected from formalin-fixed, paraffin-embedded sections to enrich DNA for regions with at least 20% cancer cell nuclei. DNA was extracted using the QIAamp DNA micro kit. At least 50 ng of DNA was used for library preparation. Amplification and adaptor ligation were performed according to manufacturer protocol. The sequences of 2 × 150 bp paired-end reads were produced from a MiSeq sequencer (Illumina, San Diego, CA, USA) following the instructions of the manufacturer. The mean coverage of each sample was >1000×. Somatic mutations were detected using the smCounter workflow. All detected variants with a minimum coverage depth of 100 reads were filtered for known single nucleotide polymorphisms (SNPs) using the Exome Sequencing Project databases and annotated by searching the Variant Effect Predictor (https://asia.ensembl.org/info/docs/tools/vep/index.html). A subsequent search in the COSMIC catalog was performed to identify driver mutations. All missense variants not reported somatic were confirmed by the Sanger sequencing of the nontumorous part.
Immunohistochemistry
Immunohistochemical staining for mucin 1 (MUC1), mucin 2 (MUC2), and β-catenin was performed on a Ventana BenchMark XT autostainer (Roche, Basel, Switzerland). After deparaffinization, rehydration, and antigen retrieval by CC1 (pH 8.0) for 20 min at 100 °C, the slides were incubated with primary antibodies for 16 min at 37 °C followed by visualization with the OptiView DAB IHC Detection Kit (Roche). The primary antibodies used are CK7 (1:50; Dako Cytomation, Carpinteria, CA, USA), CK20 (1:50; Dako Cytomation), CDX2 (1:100; Dako Cytomation), MUC1 (clone SPM493, 1:100; Thermo Fisher Scientific, Fremont, CA, USA), (clone CCP58, 1:200; Leica Biosystems, Newcastle, UK), and β-catenin (clone 14, 1:100, BD Bioscience, San Jose, CA, USA). Except for β-catenin, each marker was scored in a dichotomous manner, either showing no or low expression (≤50%) or high expression (>50%) in the tumor. β-catenin staining was classified into membranous staining only or membranous and nuclear staining.
Statistical analysis
Statistical analyses were conducted using SPSS® 19.0 software (IBM, Armonk, NY, USA). Comparisons of categorical variables were performed using the Pearson χ2 method or Fisher’s exact test when appropriate. Continuous variables were analyzed using the Student’s t test. A hierarchical cluster analysis algorithm was employed to stratify the clinicopathological features in IPNBs. All statistical results were considered significant at P < 0.05.
Results
Clinical information
The 37 patients included 20 men and 17 women. The mean age was 64.8 years (range: 35–80 year). Ten patients (27%) had stones in the biliary tracts. Sixteen tumors were located in the extrahepatic bile duct, 12 in the hilar area, and nine in the intrahepatic bile duct. Macroscopic mucin hypersecretion was identified in 11 patients. Operative methods were as follows: lobectomy in 15, atypical hepatectomy (resection not based on the segmental anatomy) in 4, wedge resection of liver in 1, partial resection of bile duct in 6, and Whipple operation in 11 cases.
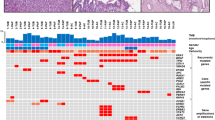
Morphologically, 18 tumors were classified as intestinal type, 2 as gastric type, 15 as pancreatobiliary type, and 2 as oncocytic type. According to the Japan–Korea consensus classification, 11 belonged to type 1 and 26 belonged to type 2 (Fig. 1). Foci of high-grade dysplasia were identified in 22 tumors. Twenty-five tumors had invasive components (Table 1).

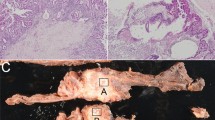
Representative morphology of intraductal papillary neoplasm of the bile duct (IPNB). a Intestinal-type IPNB, also classified as a Japan–Korea consensus type 1 IPNB based on its similarity to pancreatic intestinal type IPMN. b Intestinal-type IPNB, but classified as a Japan–Korea consensus type 2 IPNB. c Pancreatobiliary-type IPNB with focal stromal invasion. d Oncocytic-type IPNB. b–d Classified as Japan–Korea consensus type 2 IPNBs because of their complex histological architecture with irregular papillary branching or foci of solid or tubular components
Identification of frequently mutated genes
Parallel sequencing was performed for all 37 tumors. Of the 11 genes analyzed, 6 genes displayed mutations in more than 10% of all cases (Fig. 2). The most frequently mutated gene was KRAS (18 of 37, 48.6%). All mutations were nucleotide substitutions in codons 12, 13, and 61. GNAS mutations were identified in 12 tumors, resulting in the replacement of arginine residue by either histidine or cysteine in codon 201. APC and RNF43 were mutated in nine tumors. Most mutations in these two tumor suppressor genes were frameshift or nonsense mutations. Six APC mutations (67%) were located in the β-catenin binding domain. Nine tumors harbored TP53 mutations, mostly common missense mutations in the DNA-binding domains. Mutations in CTNNB1, the gene encoding the β-catenin protein, were identified in four tumors. All these mutations were known activating mutations in the GSK3 phosphorylation sites (p.S37F, p.S37A, p.T41A, and p.S45A). Mutations in three Wnt pathway genes (APC, RNF43, and CTNNB1) were mutually exclusive.

a The case numbers of IPNB with mutations of 9 cancer-related genes. b–d The distribution of somatic mutations of RNF43 (b), APC (c), and TP53 (d) in 37 cases of IPNB
Correlation of mutated genes with clinicopathologic features
We analyzed the clinicopathologic features of tumors with mutations in the six genes (Supplementary Tables 2 and 3). GNAS mutations were associated with a larger tumor size, and the presence of gross mucin. Histologically, GNAS mutations were strongly associated with the intestinal subtype, the Japan–Korea consensus type 1, and an absence of high-grade dysplasia. GNAS mutations often coexisted with KRAS and RNF43 mutations. RNF43 mutations were associated with gross mucin and the intestinal type. KRAS mutations were more frequently observed in the intestinal subtype and in the Japan–Korea consensus type 1.
Seven of the nine APC-mutated tumors and all four CTNNB1-mutated tumors were located in the extrahepatic bile duct. Morphologically, all four tumors with CTNNB1 mutations were classified as the pancreatobiliary subtype and Japan–Korea consensus type 2.
Nuclear expression of β-catenin in IPNB correlated with mutations in Wnt/β-catenin pathway genes
Because 22 tumors (59%) had mutations in Wnt/β-catenin pathway genes (APC, RNF43, and CΤΝΝΒ1), we studied the expression pattern of β-catenin by immunohistochemistry. Most tumors revealed predominant membranous/cytoplasmic β-catenin expressions with an occasional weak nuclear expression of β-catenin. Eight tumors exhibited a strong nuclear expression of β-catenin. All eight tumors harbored mutations in the Wnt/β-catenin pathway genes (CTNNB1: 4, APC: 2, and RNF43: 2) (Fig. 3).

a, b A case of intestinal type, Japan–Korea consensus type 1 IPNB shows membranous staining of β-catenin (b). c, d A case of pancreatobiliary type, Japan–Korea consensus type 2 IPNB with CΤΝΝΒ1 mutation shows nuclear localization of β-catenin in groups of tumor cells (d)
Clinicopathologic and molecular features of cell lineage subtypes and Japan–Korea consensus classification
Our previous studies have demonstrated that intestinal-type IPNB had distinct clinicopathologic and molecular features [4, 7]. Therefore, we stratified the 37 tumors according to the cell lineage subtype and the newly proposed Japan–Korea consensus classification. In our cohort, 18 tumors were classified as the intestinal subtype and 11 tumors were classified as the Japan–Korea consensus type 1. All 11 Japan–Korea consensus type 1 tumors were intestinal-type tumors. Intestinal type is associated with older age, presence of gross mucin, and absence of high-grade dysplasia. As expected, most intestinal-type IPNBs displayed a strong expression of intestinal markers CK20, MUC2, and CDX2. KRAS, GNAS, and RNF43 were more frequently mutated in intestinal-type IPNBs.
Japan–Korea consensus type 1 IPNB was also associated with older age, presence of gross mucin, and absence of high-grade dysplasia, strong expression of intestinal markers, and KRAS and GNAS mutations.
Cluster analysis of IPNB
We applied hierarchical cluster analysis to subclassify the 37 IPNBs and identify the key factor to determine the classification (Fig. 4). Ten cases were tightly clustered (group 1). They were characterized by intestinal differentiation, Japan–Korea consensus type 1, macroscopic mucin, age > 60 years, KRAS, GNAS, and RNF43 mutations. A second group was mainly composed of cases with intestinal differentiation but classified as Japan–Korea consensus type 2 in terms of histological architecture (group 2). Tumors belonging to this group were also frequently KRAS-mutated but they rarely had GNAS mutation, MUC2 expression, or macroscopic mucin. Among the remaining cases, the four CTNNB1-mutated cases were clustered in one last group (group 3). They were characterized by nuclear expression of β-catenin, extrahepatic location, lack of expression of intestinal markers, Japan–Korea consensus type 2, and lack of mutations in KRAS, APC, RNF43, and GNAS.

Hierarchical clustering of IPNB according to the clinicopathologic and molecular features. Three groups were identified. Group 1: Japan–Korea consensus type 1. Group 2: Japan–Korea consensus type 2 intestinal type. Group 3: CTNNB1-mutated
Discussion
In this study, we identified six genes that are frequently encountered in IPNB. KRAS is mutated in nearly half of all cases. Activating mutations in the KRAS genes result in the constitutive activation of the MAPK pathway [10]. RNF43 encodes an E3 ubiquitin ligase, which induces Frizzled ubiquitination and suppresses the Wnt signaling pathway [11]. Apart from the known RNF43 mutations, we identified mutations in two other Wnt/β-catenin pathway genes, APC, and CTNNB1. In total, 22 tumors (59%) had mutations in Wnt/β-catenin pathway genes. Another frequently mutated gene, GNAS, encodes the subunit of stimulatory guanine nucleotide-binding protein (Gsa), which regulates the adenylate cyclase activity upon activation of GPCRs [12]. These results indicated the Ras-MAPK, Wnt/β-catenin, and GPCR-cAMP signaling were the main drivers of IPNB.
IPNB is viewed as the biliary counterpart of IPMN. Some molecular events, such as mutations in KRAS, GNAS, and RNF43, are shared by both types of tumors. However, during the analysis of 169 genes commonly mutated in cancer, APC, CTNNB1, and TP53 mutations were not identified in IPMN [13]. Springer et al. analyzed 96 IPMNs and discovered mutations in APC and CTNNB1 in only 0% and 6% of IPMN, respectively [14]. These results suggest a different molecular pathogenesis between IPNB and IPMN. Morphologically, we have established that, unlike IPMNs, many IPNBs cannot simply be classified into a gastric type, pancreatobiliary type, or oncocytic type. These tumors tend to have a more complex architecture with papillary or tubulocystic configuration. High-grade dysplasia is frequently identified in this subgroup. Fujikura et al. proposed the term “papillary cholangiocarcinoma” to describe the cases with complex histologic architecture [15]. Recently, a group of Japanese and Korean expert pathologists proposed that IPNB be separated in two types. Type 1 IPNB is a histologic counterpart of IPMN and typically develops in the intrahepatic bile ducts. Type 2 IPNB has a more complex histological architecture and typically involves the extrahepatic bile ducts. Our study concurs with this classification in several aspects. We observed that type 1 IPNB more frequently harbored KRAS, GNAS, and RNF43 mutations than type 2 IPNB, suggesting that besides morphology, type 1 IPNB is molecularly similar to IPMN. We also discovered type 2 IPNB was more likely to have foci of high-grade dysplasia. However, we found subtle differences between our results and theirs. All type 1 IPNBs in our study were classified as the intestinal type whereas their report indicated most type 1 IPNBs were classified as gastric type. We did not detect a relevant association between tumor types and locations. The discrepancy may be due to different criteria in assigning histologic subtypes. Some tumors have mixed cell lineages. We tended to classify tumors with both gastric-type and intestinal-type epithelium as intestinal type because intestinal epithelium with expression of intestinal markers is usually the major component of these tumors. Another explanation for these discrepancies might be the true epidemiologic differences in between countries. In Taiwan, numerous IBNBs arise from a background of cholangiopathy, mainly due to hepatolithiasis (4). IPNBs associated with hepatolithiasis are more likely to be of the intestinal type with expressions of CDX2 and MUC2, and gross mucin production [16, 17]. Additional cross-country studies should be conducted to clarify this aspect.
During preparation of this report, Fujikura et al. reported mutations in APC or CTNNB1 in 6 out of 14 IPNBs (type 1 IPNB in the Japan–Korea consensus classification) [18]. However, none of the mutations were identified in intraductal cholangiocarcinoma (type 2 IPNB in the Japan–Korea classification). Our results demonstrated APC or CTNNB1 mutations were more frequently identified in type 2 IPNB. Furthermore, they did not detect any GNAS and RNF43 mutations in the 14 type 1 and 14 type 2 cases. Another Japanese group reported GNAS mutations in 50% of IPNBs [8], but unlike our study, only 1 of 30 tumors in their study was classified as the intestinal subtype. GNAS mutation did not correlate with clinicopathologic features in their study. These results further suggest differences in the mass-forming tumors of the bile duct depending on the countries.
All four IPNBs with CTNNB1 mutations were pancreatobiliary-type tumors located in the distal extrahepatic bile duct. As the case number was too small, we are not able to confirm whether these tumors constitute a distinct subtype of IPNB. CTNNB1 mutations were frequently detected in gallbladder adenoma [19]. This group of IPNBs may be the bile duct counterpart of gallbladder adenoma.
Including only 11 genes in our analysis might represent a substantial impediment. Fujikura et al. identified mutations in CDC27, histone methyltransferases (KMT2C and KMT2D), and DNA mismatch repair proteins (MSH3, MSH6, and PMS2) in one patient each. A larger cohort of whole-exome sequencing studies is required to demonstrate these genes are frequently mutated in IPNB and are functionally relevant.
Intraductal tubulopapillary neoplasms (ITPNs) are another type of intraductal mass-forming epithelial neoplasm in the pancreas and biliary tree. These tumors are characterized by a tubular architecture and the absence of or minimal mucin production. Unlike IPNBs and IPMNs, the intestinal markers CDX2 and MUC2 are not expressed in ITPNs [20,21,22]. A molecular study indicated that the loss of CDKN2A/p16 expression and/or loss of heterozygosity of the CDKN2A locus were frequent events in biliary ITPNs (intraductal components = 44%, invasive = 33%). Furthermore, TP53 mutations were identified in 17% of intraductal components and 9% of invasive tumors, and mutations in KRAS, GNAS, and PIK3CA were rare or absent. Moreover, the loss of the CDKN2A locus and lack of mutations in the Ras-MAPK, Wnt/β-catenin, and GPCR-cAMP signaling pathways were identified in pancreatic ITPNs [22]. In addition, pancreatic ITPNs frequently harbored mutations in the phosphatidylinositol 3-kinase pathway (PIK3CA, PIK3CB, INPP4A, and PTEN) and FGFR2 fusions [22]. In the present study, we did not identify PIK3CA mutations in our cohort, and the differences in the mutated genes suggested that IPNBs and ITPNs are distinct neoplasms with different molecular features.
In the pancreas, oncocytic-type IPMNs are increasingly been recognized as a distinct entity. Whole-genome sequencing of nine cases revealed the absence of GNAS and KRAS mutations in typical oncocytic IPMNs. By contrast, intestinal-subtype IPMNs with focal oncocytic epithelia frequently had TP53, GNAS, and RNF43 mutations [23]. Our study only included two oncocytic IPNBs; one had mutations in APC (p.Thr1556fs) and KRAS (p.Gln61His), whereas the other had mutations in TP53 (p.Pro152Leu). Further reviewing the slides revealed that these two tumors were not entirely composed of oncocytic cells; pancreatobiliary-type cells were identified in both tumors. Therefore, these two tumors may not be the biliary counterparts of typical oncocytic IPNBs of the pancreas, and further studies of more cases are required for clarification.
In conclusion, we identified frequent mutations in six tumor-related genes in IPNB. The results indicate Ras-MAPK, Wnt/β-catenin, and GPCR/cAMP signaling are the main drivers of IPNB. We confirmed that KRAS, GNAS, and RNF43 mutations are closely associated with the intestinal type and Japan–Korea consensus type 1, supporting the concept that IPNB is a heterogeneous neoplasm and that further classification is mandatory. The presence of APC and CTNNB1 mutations suggest that at least a subset of IPNB is molecularly distinct to pancreatic IPMN.
References
Naito Y, Kusano H, Nakashima O, et al. Intraductal neoplasms of the intrahepatic bile duct: clinicopathological study of 24 cases. World J Gastroenterol. 2012;18:3673–80.
Bickenbach K, Galka E, Roggin KK. Molecular mechanisms of cholangiocarcinogenesis: are biliary intraepithelial neoplasia and intraductal papillary neoplasms of the bile duct precursors to cholangiocarcinoma? Surg Oncol Clin N Am. 2009;18:215–24.
Nakanuma Y, Uesaka K, Miyayama S, et al. Intraductal neoplasms of the bile duct. A new challenge to biliary tract tumor pathology. Histol Histopathol. 2017;32:1001–15.
Tsai JH, Yuan RH, Chen YL, et al. GNAS is frequently mutated in a specific subgroup of intraductal papillary neoplasms of the bile duct. Am J Surg Pathol. 2013;37:1862–70.
Zen Y, Fujii T, Itatsu K, et al. Biliary papillary tumors share pathological features with intraductal papillary mucinous neoplasm of the pancreas. Hepatology. 2006;44:1333–43.
Nakanuma Y, Jang KT, Fukushima N, et al. A statement by the Japan-Korea expert pathologists for future clinicopathological and molecular analyses toward consensus building of intraductal papillary neoplasm of the bile duct through several opinions at the present stage. Hepatobiliary Pancreat Sci. 2018;25:181–7.
Tsai JH, Liau JY, Yuan CT, et al. RNF43 mutation frequently occurs with GNAS mutation and mucin hypersecretion in intraductal papillary neoplasms of the bile duct. Histopathology. 2017;70:756–65.
Sasaki M, Matsubara T, Nitta T, et al. GNAS and KRAS mutations are common in intraductal papillary neoplasms of the bile duct. PLoS ONE. 2013;8:e81706.
Schlitter AM, Born D, Bettstetter M, et al. Intraductal papillary neoplasms of the bile duct: stepwise progression to carcinoma involves common molecular pathways. Mod Pathol. 2014;27:73–86.
McCormick F. KRAS as a therapeutic target. Clin Cancer Res. 2015;21:1797–801.
Koo BK, Spit M, Jordens I, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012;488:665–9.
Weinstein LS, Liu J, Sakamoto A, et al. Minireview: GNAS: normal and abnormal functions. Endocrinology. 2004;145:5459–64.
Wu J, Matthaei H, Maitra A, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011;3:92ra66.
Springer S, Wang Y, Dal Molin M, et al. A combination of molecular markers and clinical features improve the classification of pancreatic cysts. Gastroenterology. 2015;149:1501–10.
Fujikura K, Fukumoto T, Ajiki T, et al. Comparative clinicopathological study of biliary intraductal papillary neoplasms and papillary cholangiocarcinomas. Histopathology. 2016;69:950–61.
Ishikawa A, Sasaki M, Ohira S, et al. Aberrant expression of CDX2 is closely related to the intestinal metaplasia and MUC2 expression in intraductal papillary neoplasm of the liver in hepatolithiasis. Lab Invest. 2004;84:629–38.
Bae JY, Park YN, Nakanuma Y, et al. Intestinal type cholangiocarcinoma of intrahepatic large bile duct associated with hepatolithiasis–a new histologic subtype for further investigation. Hepatogastroenterology. 2002;49:628–30.
Fujikura K, Akita M, Ajiki T, et al. Recurrent mutations in APC and CTNNB1 and activated Wnt/β-catenin signaling in intraductal papillary neoplasms of the bile duct: a whole exome sequencing study. Am J Surg Pathol. 2018;42:1674–85.
Chang HJ, Jee CD, Kim WH. Mutation and altered expression of β-catenin during gallbladder carcinogenesis. Am J Surg Pathol. 2002;26:758–66.
Schlitter AM, Jang KT, Klöppel G, et al. Intraductal tubulopapillary neoplasms of the bile ducts: clinicopathologic, immunohistochemical, and molecular analysis of 20 cases. Mod Pathol. 2015;28:1249–64.
Basturk O, Adsay V, Askan G, et al. Intraductal tubulopapillary neoplasm of the pancreas: a clinicopathologic and immunohistochemical analysis of 33 cases. Am J Surg Pathol. 2017;41:313–25.
Basturk O, Berger MF, Yamaguchi H, et al. Pancreatic intraductal tubulopapillary neoplasm is genetically distinct from intraductal papillary mucinous neoplasm and ductal adenocarcinoma. Mod Pathol. 2017;30:1760–72.
Basturk O, Tan M, Bhanot U, et al. The oncocytic subtype is genetically distinct from other pancreatic intraductal papillary mucinous neoplasm subtypes. Mod Pathol. 2016;29:1058–69.
Acknowledgements
This research is supported by Ministry of Science and Technology, Republic of China (Grant number 107-2320-B-002-047) and Taipei City Hospital.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Yang, CY., Huang, WJ., Tsai, JH. et al. Targeted next-generation sequencing identifies distinct clinicopathologic and molecular entities of intraductal papillary neoplasms of the bile duct. Mod Pathol 32, 1637–1645 (2019). https://doi.org/10.1038/s41379-019-0306-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0306-9
This article is cited by
-
Intraductal oncocytic papillary neoplasm arising in Peutz-Jeghers Syndrome bile duct: a unique case report
Diagnostic Pathology (2022)
-
Spatiotemporal analysis of tumour-infiltrating immune cells in biliary carcinogenesis
British Journal of Cancer (2022)
-
Whole-exome sequencing reveals the etiology of the rare primary hepatic mucoepidermoid carcinoma
Diagnostic Pathology (2021)
-
Intracholecystic papillary neoplasm arising in the cystic duct and extending into common bile duct: a case report
Clinical Journal of Gastroenterology (2021)
