
Abstract
To systematically evaluate genetic susceptibility to type 2 diabetes (T2D) in “candidate” regions on chromosomes 1q, 3q and 12q, we examined disease association by using a total of 2,083 SNPs in two-step screening; a screening panel comprised 473 cases and 285 controls and an extended (or combined) panel involved 658 cases and 474 controls. For the total interval screened (40.9 Mb), suggestive evidence of association was provided for several annotated gene loci. For example, in the MCF2L2 gene on 3q, a significant association (a nominal P value of 0.00009) was observed when logistic regression analysis was performed for three associated SNPs (rs684846, rs35069869 and rs35368790) that belonged to different LD groups. Also, in the SLC15A4 gene on 12q, rs3765108 showed a marginally significant association with an overall estimated odds ratio of 0.79 (P = 0.001). No significant association was found for known candidate gene loci on 3q, such as ADIPOQ and IGF2BP2. Using the available samples, we have observed disease associations of SNPs derived from two novel gene loci in the Japanese population through high-density searches of diabetes susceptibility in three chromosomal regions. Independent replication will clarify the etiological relevance of these genomic loci to T2D.
Similar content being viewed by others

Introduction
Considerable efforts have been made in the study of the molecular genetics of type 2 diabetes (T2D), but the inherently complex nature of the task has hampered progress in the elucidation of the genes involved (Florez et al. 2003). Over the last decade, multiple genome-wide linkage analyses have been conducted by using microsatellite markers to localize genes influencing diabetes status and/or associated metabolic trait phenotypes in a number of populations derived from various ethnic groups. While few single studies have so far yielded definitive evidence for “principal” diabetes susceptibility gene(s), some of these studies provide consistency of linkage results in several chromosomal regions (Stern 2002). Peaks of linkage have been mapped to a relatively broad interval (>10–20 Mb) and the challenge of narrowing these regions to facilitate gene discovery remains formidable. Nevertheless, the linkages that have been replicated thus far provide good starting points to explore functional variants in T2D susceptibility genes.
On chromosomes 1q, 3q and 12q, suggestive evidence of linkage to T2D has been repeatedly documented in several ethnic groups. Here, it is worth noting that replications have been reported in at least two different ethnic groups even when linkages are limited to phenotypes related directly to the trait of diabetes, i.e., when those related to obesity or other metabolic variables are not considered. Also, these three regions are known to harbor positional candidate genes of physiological importance. On chromosome 1q, significant linkage has been reported in Caucasian (Elbein et al. 1999; Vionnet et al. 2000; Wiltshire et al. 2001), Pima Indian (Hanson et al. 1998) and Chinese (Ng et al. 2004) populations, with the LOD score peaks being located in an interval between 166 and 186 cM (according to the Marshfield map, http://research.marshfieldclinic.org/genetics/), near the apolipoprotein A-II (APOA2) gene. On chromosome 3q, significant linkage has been also reported in Caucasian (Vionnet et al. 2000), Native American (Oji-Cree) (Hegele et al. 1999) and Japanese (Mori et al. 2002) populations, with the LOD score peaks being located in an interval between 214 and 217 cM, near the adiponectin (ADIPOQ) gene. Furthermore, on chromosome 12q, significant linkage has been reported in Caucasian (Mahtani et al. 1996; Shaw et al. 1998; Bowden et al. 1997; Ehm et al. 2000) and African-American (Ehm et al. 2000) populations, with the LOD score peaks being located in an interval between 135 and 145 cM, near the HNF-1α, or tissue factor 1 (TCF1) gene. Within the three regions, IGF2BP2 on chromosome 3q was reported to confer diabetes susceptibility in a recent whole-genome association study in Caucasians (Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes for BioMedical Research 2007; Zeggini et al. 2007; Scott et al. 2007).
To systematically evaluate genetic susceptibility to T2D in the relevant regions, we performed an extensive “candidate region” approach with a case-control association study on chromosomes 1q, 3q and 12q, where linkage to T2D had been identified through several genome-wide searches with substantial overlap (Stern 2002).
Subjects and methods
Study population and marker genotyping
Participants were recruited from the patients and their spouses who came regularly to the outpatient clinic of the Hospital of International Medical Center of Japan in Tokyo and the Hiranuma Clinic in Yokohama, both of which are located in the metropolitan area of Japan. The diagnosis of T2D was based on the criteria of the World Health Organization. The patients with secondary diabetic disorders and maturity onset diabetes of the young were excluded. The normal control subjects were selected according to the following criteria: no past history of urinary glucose or glucose intolerance, an HbA1c level <5.6% or a normal glucose (75g) tolerance test, and age >60 years. Thus, a total of 658 cases and 474 controls were enrolled in the present study. All study subjects were unrelated, and they gave written consent for participation after being informed of the purpose of the study. The study protocol was approved by the ethics committee of the International Medical Center of Japan.
The SNPs for genotyping were initially selected from the TaqMan® SNP Genotyping Assays (Applied Biosystems, Foster City, CA, USA) in the candidate regions for T2D on chromosomes 1q, 3q and 12q. We focused on the 1q region spanning a 6.7-Mb interval (from D1S398 to D1S2878), the 3q region spanning a 19.1-Mb interval (from D3S1565 to D3S1601) and the 12q region spanning a 15.2-Mb interval (from D12S369 to D12S1714), each of which corresponded to a LOD-1 interval from the previous studies showing significant linkages (Stern 2002). While some were excluded due to a low minor allele frequency (MAF) in the Japanese, i.e., <0.05, a consecutive set of the TaqMan® Assays were used for the screening of diabetes susceptibility gene(s) on each chromosome. Also, in several regions where appropriate SNPs were unavailable from the TaqMan® Assays, we sought polymorphic markers from the dbSNP or JSNP (Japanese SNP) databases alternatively. Two-step screening was performed as follows. First, all of the aforementioned SNPs were genotyped in a screening panel comprising 473 cases and 285 controls (Table 1). Second, after evaluating the statistical significance by using two test statistics (the MAX trend test for genotype distributions described below and independence on [2 × 2] contingency table for allele frequencies), SNPs that showed significant (P < 0.01) differences in either of the tests were further examined in a combined panel involving an additional panel of 185 cases and 189 controls plus the screening panel. Subsequent to the initial screening, as few significant SNPs within annotated genes could be detected in the 1q and 12q regions studied, the threshold was relaxed from P < 0.01 to P < 0.05 to pick up relatively modest associations. In order to include the gaps within 10 kb from the SNPs that showed modestly significant (P < 0.05) associations in the initial screening, we genotyped additional SNPs when applicable. A total of 2,083 SNPs with MAF ≥ 0.05 were genotyped; 396 SNPs in a 6.7-Mb interval on 1q (an adjacent marker interval was 16.9 ± 1.1 kb), 948 SNPs in a 19.1-Mb interval on 3q (an adjacent marker interval was 20.2 ± 0.7 kb) and 739 SNPs in a 15.2-Mb interval on 12q (an adjacent marker interval was 20.5 ± 0.9 kb). This set of SNPs covered a total of 407 genes (120 on 1q, 143 on 3q and 144 on 12q) that were annotated in the relevant chromosomal regions (NCBI Build 36.2). The arbitrary threshold of statistical significance in the combined panel was set to be P < 0.01 in either of the two tests.
In the ADIPOQ and IGF2BP2 gene regions, we genotyped a number of SNPs including those that had previously shown significant association with T2D (Scott et al. 2007; Rodriguez et al. 2007; Hara et al. 2002; Nakatani et al. 2005).
Statistical analysis
Association analysis
The SNPs were tested individually for the statistical significance of disease association by using the χ 2-test statistic for the allele-based test and the MAX trend test (the maximum of the standardized version of the three tests optimal for additive, dominant and recessive modes of inheritance, which is robust and powerful when the underlying genetic model is unknown) with the P value computed by permutation for the genotype-based test (Freidlin et al. 2002). SNP genotype departures from Hardy–Weinberg equilibrium (HWE) were tested using a χ 2-test with one degree of freedom with a cut-off P value of 0.0001 for cases and 0.001 for controls. The extent of linkage disequilibrium (LD) was measured in terms of an LD coefficient r 2 before the analysis of haplotype structure. By categorizing any pair of SNPs having r 2 ≥ 0.6 (but not necessarily adjacent on the chromosome) into the same LD group, we defined LD groups along the chromosomal regions studied. Within each LD group, haplotypes were inferred from genotype data by the SNPHAP software (http://archimedes.well.ox.ac.uk/pise/snphap-simple.html) jointly for the cases and controls. This restriction of SNPs to those constituting individual LD groups could enhance the power to detect a disease locus (Takeuchi et al. 2005). Haplotype class counts for the separate case and control samples were then calculated, where haplotypes with frequencies less than 1% were neglected.
Genetic analysis in the selected candidates
Suggestive evidence of association was detected in the initial screening around NOS1AP [nitric oxide synthase 1 (neuronal) adaptor protein] on 1q, and MCF2L2 [MCF.2 cell line derived transforming sequence-like 2 and the interleukin 1 receptor accessory protein] and IL1RAP [interleukin 1 receptor accessory protein] on 3q, and additionally SLC15A4 [solute carrier family 15, member 4], RNF34 [ring finger protein 34] and AACS [acetoacetyl-CoA synthetase] on 12q. Accordingly, LD relations of SNPs were further examined in detail as previously reported (Takeuchi et al. 2005).
Calculation of false discovery rate
To account for multiple testing of 2,083 SNPs, we evaluated the false discovery rate (FDR), a practical and powerful approach to multiple testing (Benjamini and Hochberg 1995), at P values observed for individual genetic markers separately in individual chromosomal regions, which each have prior evidence as shown by linkage scans. The FDR is determined from the observed P value distribution to provide a measure of the expected proportion of false positives among the data. The FDR differs from the P value and higher values can be tolerated for FDRs than for P values in some contexts.
Results
Association study
A total of 658 diabetic patients and 474 control subjects were enrolled in the present study. The clinical characteristics of the study panel are described in Table 1. Overall success rate and accuracy of the genotyping assay exceeded 95 and 99.9%, respectively. In some instances (less than 5%), where genotyping data were not clearly distinguished in the scatter plot of the TaqMan® Assays or were not consistent with HWE, we sought alternative SNPs for the replacement. After the placement of additional SNP markers in the regions where suggestive association (P < 0.05) was detected in the initial SNP marker set, among a total of 2,083 SNPs we found two SNPs on 1q, 19 SNPs on 3q and ten SNPs on 12q to be significantly (P < 0.01 in either the genotype-based or allele-based test) associated with T2D in the initial screening (“Electronic supplementary material,” Fig. S1). These 31 SNPs and 49 SNPs that were additionally selected with the relaxed threshold (P < 0.05) on 1q and 12q, and the 110 SNPs that were located in the regions of interest, were further genotyped in the additional panel. Since we had adopted relatively generous criteria for screening of association signals, we estimated type I error probability for the two-step screening and then evaluated FDR to account for multiple testing in the entire screening. FDR was 1.73 on 1q (n = 1), 0.80 on 3q (n = 8) and 0.51 on 12q (n = 15) for the SNPs showing significant association, respectively. Here, in each chromosomal region, FDR was calculated for a set of SNPs (the number of SNPs depicted in the parentheses above) showing P < 0.01 in the combined panel and a concordant tendency for odds ratio (OR) in the screening and additional panels (Table 2).
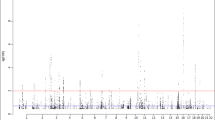
On chromosome 3q, we observed the strongest evidence for association in the MCF2L2 region (Fig. 1A) but not in either the ADIPOQ (Fig. 1C) or the IGF2BP2 regions in our studied population. Five SNPs spanning a 134-kb interval showed P = 0.002∼0.008 in MCF2L2 (Table 2 and “Electronic supplementary material” Table S1A), whereas four SNPs in ADIPOQ and one SNP in IGF2BP2 that was previously reported (Scott et al. 2007; Rodriguez et al. 2007; Hara et al. 2002; Nakatani et al. 2005) failed to show significant association in the combined panel. On chromosome 12q, we observed significant evidence for association in the SLC15A4 region (Fig. 1B); nine SNPs spanning a 27-kb interval showed P = 0.001–0.007 in SLC15A4 (Table 2 and “ESM” Table S1B). We examined disease association in nine other regions where significant P values (P < 0.01) were detected in the combined panel (Table 2). Despite detailed investigation of the neighboring SNPs, no strong evidence in favor of diabetes susceptibility genes was obtained in any of these regions (“ESM” Tables S1C, S1D, S1E and S1F and “ESM” Figs. S1 and S2).

Association of markers from the MCF2L2, SLC15A4 and ADIPOQ regions with T2D. Results for SNPs genotyped in the screening and combined panels are shown with open circles and filled circles, respectively. Horizontal dotted lines indicate a −log10P value = 2, i.e., P = 0.01. A −log10P values of the differences in allele frequencies between case and control subjects were plotted against physical distance, as centered by the marker rs35069869 in MCF2L2. At the bottom, partial exons of known and predicted genes are shown. B −log10P values of the differences in allele frequencies between case and control subjects were plotted against physical distance, as centered by the most significantly associated marker rs3765108 in SLC15A4. C −log10P values of the differences in allele frequencies between case and control subjects were plotted against physical distance, as centered by the translation initiation site of ADIPOQ
We further performed haplotype analysis by defining LD groups along the chromosomal regions and a total of 353 LD groups (which comprised more than one SNP with MAF ≥ 0.05)—72 on 1q, 175 on 3q and 106 on 12q—were constructed at the level of r 2 ≥ 0.6. None of the LD groups had P values larger than the Bonferroni correction of 0.05, which is divided by the number of groups in the individual regions (data not shown).
Genetic analysis in the MCF2L2 and SLC15A4 genes
We tested disease association with a total of 39 SNPs including additional 12 tag SNPs within the MCF2L2 gene that consisted of 36 introns spanning 250-kb on chromosome 3q. We found five SNPs—rs670232, rs684846, rs6783373, rs35069869 and rs35368790—to be significantly (P < 0.01) associated with T2D in the combined panel (Table 2). The P values for significant SNPs were similar between the ones calculated with the allele frequency and the ones calculated by using logistic regression to test the additive model with the adjustment for sex and BMI (“ESM” Table S2). According to the pattern of LD, there were five LD blocks in MCF2L2 (Fig. 2). The five significant SNPs belonged to three distinct LD blocks, and the significant SNPs from different blocks were in linkage equilibrium (r 2 < 0.05). There was no haplotype class showing more significant disease association than individual SNPs (data not shown). Also, to test the independence of multiple associated SNPs in MCF2L2, we performed logistic regression analysis by adjusting for sex and BMI as shown in Table 3. Notably, three associated SNPs that belonged to different LD blocks—rs684846, rs35069869 and rs35368790—appeared to increase the risk of being diabetic in a multiplicative manner; i.e., the model fitness involving the three SNPs was significant with the χ 2-test statistic even when the effects of multiple testing in the chromosomal region were adjusted for by permutation (a nominal P = 0.00009 and an adjusted P = 0.014) (Table 3).

LD relations between SNPs in the MCF2L2 and SLC15A4 genes (top) and disease association of markers from the corresponding genomic regions (bottom). At the top, the LD between a pair of markers is indicated by the color of the block above and to the left of the intersection of the markers. To enhance readability, only the names of SNPs showing significant association are shown to the right of the vertical axis of the LD plot. The rest of the SNP information is described in the “ESM” (Table S1A and B). At the bottom, the locations of genetic markers studied in the corresponding genomic regions are shown in relation to gene structure
Likewise, we tested disease association with a total of 18 SNPs including an additional nine tagged SNPs within the SLC15A4 gene that consisted of 12 introns spanning 30.8-kb on chromosome 12q. We found nine SNPs to be significantly (P < 0.01) associated with T2D in the combined panel (Table 2). According to the pattern of LD, there appeared to be a single LD block in SLC15A4 (Fig. 2). The SNP rs3765108 was most significant (P = 0.001) and other SNPs did not show additional contributions to disease association. Associations with nearby SNPs in strong LD were observed in this gene but not in the other positional candidates (e.g., RNF34 and AACS) on 12q. Moreover, although individual study panels (screening and additional panels) are rather modest in size, three SNPs in SLC15A4—rs3765108, rs7974764 and rs900982—showed borderline association (P < 0.05) consistently in both panels (“ESM” Table S3).
We further evaluated the potential impacts of obesity (in particular, history of obesity) on the observed disease associations in the MCF2L2 and SLC15A4 loci by subgrouping diabetic subjects according to the maximum BMI (“ESM” Fig. S3). There were no apparent confounding influences of maximum BMI on disease association for the significant SNPs detected in the present study.
Discussion
Based on the linkage results from the previous studies (Stern 2002), we have explored diabetes susceptibility genes over a combined 40.9-Mb interval that contains 407 annotated genes in three chromosomal regions. At the arbitrary threshold of P < 0.01 for 3q and P < 0.05 for 1q and 12q, 3.9% of the SNPs tested in the initial screening (80 out of 2051 SNPs) turned out to be significant, and we have attempted to validate them by increasing the study subjects. In terms of statistical significance of disease association, SNPs from MCF2L2 on chromosome 3q and those from SLC15A4 on chromosome 12q are of particular interest when we analyze the data in the combined panel (Table 2).
In the MCF2L2 region on chromosome 3q, we noticed that five SNPs are significantly associated with T2D, among which two pairs of SNPs are in strong LD to each other (Fig. 2). Notably, logistic regression analysis involving rs684846, rs35069869 and rs35368790 has indicated the potential presence of allelic heterogeneity in MCF2L2 (Table 3). These results need to be carefully interpreted, but similar findings of allelic heterogeneity have been reported in the association study of CETP gene variants regarding coronary artery disease (Horne et al. 2007).
The MCF2L2 gene is considered to encode a Rho family guanine-nucleotide exchange factor, and it produces 14 different mRNAs by alternative splicing, whereas the detailed gene function remains unknown. Of note is the fact that ARHGEF11, which encodes the Rho guanine-nucleotide exchange factor 11, was analyzed as a positional candidate gene for chromosome 1q linkage in Pima Indians because of its stimulatory activity on Rho-dependent signals, such as the insulin-signaling cascade, and a variation within ARHGEF11 appeared to nominally increase the risk of T2D (Ma et al. 2007). By quantitative RT-PCR, we have found that this gene is expressed significantly in the whole brain, and modestly in the pancreas, spleen and testis in humans. However, we could not identify the homolog of MCF2L2 in mice (data not shown).
On chromosome 12q, we have also detected several genes that contain SNPs showing significant disease association. Among such genes, SLC15A4 is highlighted as a potential candidate by our fine mapping of SNPs in the relevant genes (“ESM” Fig. S1). The SLC15A4 gene encodes a peptide-histidine transporter 4, which is expected to exhibit a proton-dependent oligopeptide transport activity in various tissues, including the pancreatic islets and colon. Though speculative, this gene may play a role in the entero-insular axis or protein nutrition regulated by insulin, similar to SLC15A1 (or PEPT1), another member of the proton-dependent oligopeptide transporters (Gangopadhyay et al. 2002). Detailed investigation including independent replication of disease association will lead us to clarify the etiological relevance of SLC15A4 and the other potential candidate gene SNPs and/or haplotypes to diabetes.
On chromosome 1q, we have found a single SNP, rs4233387, in the NOS1AP gene that shows significant disease association in the combined panel. Despite our fine mapping, there was no further evidence supporting its pathophysiological involvement with the available samples.
In view of high-resolution marker genotyping, we should consider the statistical power and type I error probability; both issues are brought up by our screening strategy. First, to address the issue of statistical power, we have performed simulation analysis by calculating the ratio of times that the cut-off level (P < 0.01) of association was surpassed among 1000-times simulation data sets (see explanation in the “ESM”). For instance, when we test SNPs with an OR of 1.5 (as in the case of the rs684846 genotype of MCF2L2) for allele frequency comparison on chromosome 3q, we can expect the statistical power which represents the ratio of [true positives/(true positives + false negatives)] to be more than 64% (79% for chromosomal regions 1q and 12q) assuming a multiplicative or additive mode of inheritance, a disease allele frequency of between 0.2 and 0.8, and 10% prevalence. Next, we have estimated the sample size required for a replication study to achieve 80% statistical power at 5% type I error probability. To correct for the ascertainment bias and to overcome the “winner’s curse,” i.e., overestimation of genetic effects based on the original data, we adjusted the ORs for significant SNPs according to the algorithm previously proposed (Zollner and Pritchard 2007). The significant SNP, rs684846, in MCF2L2 showed an OR of 1.52 (Table 2) and its adjusted OR was 1.42, for which the necessary sample size was estimated to be 510 each of cases and controls. Likewise, rs3765108 in SLC15A4 showed an OR of 1.27 and an adjusted OR of 1.23, thus requiring 582 each of cases and controls. As for rs4402960 in IGF2BP2, which showed significant association in the Caucasian population, the necessary sample size was 1,619 each for cases and controls when adopting the OR of 1.14 observed in the Caucasians (Scott et al. 2007) and the associated variant frequency of 0.32 in our Japanese samples. In these estimations, we assumed a multiplicative mode of inheritance for the individual SNPs and 10% for the disease prevalence.
From a methodological point of view, however, we should bear in mind several limitations of the present study. When we consider the average marker density (1 SNP per 16.9–20.5 kb across three chromosomal regions) and the random selection procedure without utilizing LD information, there is likely to be a certain amount of common variations not captured. Across the three target regions, we have assessed the percentage of common variations that are covered by the SNPs genotyped in the present study. Approximately 75% (297 of 396, on 1q), 78% (739 of 948, on 3q) and 74% (547 of 739, on 12q) were shared with the HapMap database (Release 19), and 32% (on 1q), 38% (on 3q) and 27% (on 12q) of the HapMap SNPs (with MAF ≥ 0.05) from JPT are estimated to have a high r 2 (>0.8) to one of SNPs genotyped. The remaining SNPs not in HapMap have captured further information on variations. Second, to evaluate the type I error probability caused by multiple testing, we have calculated the FDR at P-values observed for individual genetic markers. We have found the lowest FDR of 0.76 for significant SNPs in MCF2L2 and the lowest FDR of 0.39 for those in SLC15A4, when disease association is tested in the combined panel. Thus, even though far from definitive in the present study alone, the FDR analysis indicates that our results are worth following up. The only way to identify definitive findings from false positives is to replicate the results in other studies.
Another important issue is the lack of significant evidence for disease association in either the ADIPOQ (Fig. 1C) or IGF2BP2 regions on 3q in our Japanese population, contrary to previous findings (Table 2). This does not refute the disease causality of either of these genes by itself. Based on our power calculation as mentioned above, it is possible that the underpowered sample size results in the lack of evidence for association in the ADIPOQ and IGF2BP2 regions. It is also possible that diabetes susceptibility gene(s) differ among ethnic groups, since peaks of linkage have been mapped to a relatively broad region of chromosome 3q with partial overlapping.
Alternatively, we sought disease associations of the MCF2L2 and SLC15A4 genes in the Caucasian population by looking into the public data (the Wellcome Trust Case Control Consortium, http://www.wtccc.org.uk/info/summary_stats.shtml, and the Diabetes Genetics Initiative, http://www.broad.mit.edu/diabetes/scandinavs/type2.html). Among SNPs in the publicly available data set, only one SNP (rs670232) in the MCF2L2 gene was common to the list of SNPs showing significant association in our study, and five SNPs in the SLC15A4 gene were in strong LD with our significant SNPs. However, the relevant SNPs did not show significant association in Caucasian populations (“ESM” Table S4).
In conclusion, we have identified the MCF2L2 gene on chromosome 3q as a likely positional candidate for conferring genetic susceptibility to T2D in the Japanese population. Our data also indicate that on chromosome 12q, molecular variations in the SLC15A4 gene may increase the risk of T2D. These findings warrant further replication study, but if replicated, their biological relevance will become an issue of great interest.
References
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300
Bowden DW, Sale M, Howard TD, Qadri A, Spray BJ, Rothschild CB, Akots G, Rich SS, Freedman BI (1997) Linkage of genetic markers on human chromosomes 20 and 12 to NIDDM in Caucasian sib pairs with a history of diabetic nephropathy. Diabetes 46:882–886
Diabetes Genetics Initiative of Broad Institute of Harvard MIT, Lund University, and Novartis Institutes for BioMedical Research (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316:1331–1336
Ehm MG, Karnoub MC, Sakul H, Gottschalk K, Holt DC, Weber JL, Vaske D, Briley D, Briley L, Kopf J, McMillen P, Nguyen Q, Reisman M, Lai EH, Joslyn G, Shepherd NS, Bell C, Wagner MJ, Burns DK; American Diabetes Association GENNID Study Group (2000) Genetics of NIDDM. Genomewide search for type 2 diabetes susceptibility genes in four American populations. Am J Hum Genet 66:1871–1881
Elbein SC, Hoffman MD, Teng K, Leppert MF, Hasstedt SJ (1999) A genome-wide search for type 2 diabetes susceptibility genes in Utah Caucasians. Diabetes 48:1175–1182
Florez JC, Hirschhorn J, Altshuler D (2003) The inherited basis of diabetes mellitus: implications for the genetic analysis of complex traits. Annu Rev Genomics Hum Genet 4:257–291
Freidlin B, Zheng G, Li Z, Gastwirth JL (2002) Trend tests for case-control studies of genetic markers: power, sample size and robustness. Hum Hered 53:146–152
Gangopadhyay A, Thamotharan M, Adibi SA (2002) Regulation of oligopeptide transporter (Pept-1) in experimental diabetes. Am J Physiol Gastrointest Liver Physiol 283:G133–G138
Hanson RL, Ehm MG, Pettitt DJ, Prochazka M, Thompson DB, Timberlake D, Foroud T, Kobes S, Baier L, Burns DK, Almasy L, Blangero J, Garvey WT, Bennett PH, Knowler WC (1998) An autosomal genomic scan for loci linked to type II diabetes mellitus and body-mass index in Pima Indians. Am J Hum Genet 63:1130–1138
Hara K, Boutin P, Mori Y, Tobe K, Dina C, Yasuda K, Yamauchi T, Otabe S, Okada T, Eto K, Kadowaki H, Hagura R, Akanuma Y, Yazaki Y, Nagai R, Taniyama M, Matsubara K, Yoda M, Nakano Y, Tomita M, Kimura S, Ito C, Froguel P, Kadowaki T (2002) Genetic variation in the gene encoding adiponectin is associated with an increased risk of type 2 diabetes in the Japanese population. Diabetes 51:536–540
Hegele RA, Sun F, Harris SB, Anderson C, Hanley AJ, Zinman B (1999) Genome-wide scanning for type 2 diabetes susceptibility in Canadian Oji-Cree, using 190 microsatellite markers. J Hum Genet 44:10–14
Horne BD, Camp NJ, Anderson JL, Mower CP, Clarke JL, Kolek MJ, Carlquist JF; Intermountain Heart Collaborative Study Group (2007) Multiple less common genetic variants explain the association of the cholesteryl ester transfer protein gene with coronary artery disease. J Am Coll Cardiol 49:2053–2060
Ma L, Hanson RL, Que LN, Cali AM, Fu M, Mack JL, Infante AM, Kobes S; International Type 2 Diabetes 1q Consortium, Bogardus C, Shuldiner AR, Baier LJ (2007) Variants in ARHGEF11, a candidate gene for the linkage to type 2 diabetes on chromosome 1q, are nominally associated with insulin resistance and type 2 diabetes in Pima Indians. Diabetes 56:1454–1459
Mahtani MM, Widen E, Lehto M, Thomas J, McCarthy M, Brayer J, Bryant B, Chan G, Daly M, Forsblom C, Kanninen T, Kirby A, Kruglyak L, Munnelly K, Parkkonen M, Reeve-Daly MP, Weaver A, Brettin T, Duyk G, Lander ES, Groop LC (1996) Mapping of a gene for type 2 diabetes associated with an insulin secretion defect by a genome scan in Finnish families. Nat Genet 14:90–94
Mori Y, Otabe S, Dina C, Yasuda K, Populaire C, Lecoeur C, Vatin V, Durand E, Hara K, Okada T, Tobe K, Boutin P, Kadowaki T, Froguel P (2002) Genome-wide search for type 2 diabetes in Japanese affected sib-pairs confirms susceptibility genes on 3q, 15q, and 20q and identifies two new candidate loci on 7p and 11p. Diabetes 51:1247–1255
Nakatani K, Noma K, Nishioka J, Kasai Y, Morioka K, Katsuki A, Hori Y, Yano Y, Sumida Y, Wada H, Nobori T (2005) Adiponectin gene variation associates with the increasing risk of type 2 diabetes in non-diabetic Japanese subjects. Int J Mol Med 15:173–177
Ng MC, So WY, Cox NJ, Lam VK, Cockram CS, Critchley JA, Bell GI, Chan JC (2004) Genome-wide scan for type 2 diabetes loci in Hong Kong Chinese and confirmation of a susceptibility locus on chromosome 1q21–q25. Diabetes 53:1609–1613
Rodriguez S, Gaunt TR, Day IN (2007) Molecular genetics of human growth hormone, insulin-like growth factors and their pathways in common disease. Hum Genet 122:1–21
Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M (2007) A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316:1341–1345
Shaw JT, Lovelock PK, Kesting JB, Cardinal J, Duffy D, Wainwright B, Cameron DP (1998) Novel susceptibility gene for late-onset NIDDM is localized to human chromosome 12q. Diabetes 47:1793–1796
Stern MP (2002) The search for type 2 diabetes susceptibility genes using whole-genome scans: an epidemiologist’s perspective. Diabetes Metab Res Rev 18:106–113
Takeuchi F, Yanai K, Morii T, Ishinaga Y, Taniguchi-Yanai K, Nagano S, Kato N (2005) Linkage disequilibrium grouping of SNPs reflecting haplotype phylogeny for efficient selection of tag SNPs. Genetics 170:291–304
Vionnet N, Hani EH, Dupont S, Gallina S, Francke S, Dotte S, De Matos F, Durand E, Lepretre F, Lecoeur C, Gallina P, Zekiri L, Dina C, Froguel P (2000) Genomewide search for type 2 diabetes-susceptibility genes in French whites: evidence for a novel susceptibility locus for early-onset diabetes on chromosome 3q27-qter and independent replication of a type 2-diabetes locus on chromosome 1q21-q24. Am J Hum Genet 67:1470–1480
Wiltshire S, Hattersley AT, Hitman GA, Walker M, Levy JC, Sampson M, O’Rahilly S, Frayling TM, Bell JI, Lathrop GM, Bennett A, Dhillon R, Fletcher C, Groves CJ, Jones E, Prestwich P, Simecek N, Rao PV, Wishart M, Bottazzo GF, Foxon R, Howell S, Smedley D, Cardon LR, Menzel S, McCarthy MI (2001) A genomewide scan for loci predisposing to type 2 diabetes in a U.K. population (the Diabetes UK Warren 2 Repository): analysis of 573 pedigrees provides independent replication of a susceptibility locus on chromosome 1q. Am J Hum Genet 69:553–569
Zeggini E, Weedon MN, Lindgren CM Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney AS; Wellcome Trust Case Control Consortium (WTCCC), McCarthy MI, Hattersley AT (2007) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316:1336–1341
Zollner S, Pritchard JK (2007) Overcoming the winner’s curse: estimating penetrance parameters from case-control data. Am J Hum Genet 80:605–615
Acknowledgments
This work was supported by grants from the Program for Promotion of Fundamental Studies in Health Sciences of Pharmaceuticals and Medical Devices Agency (PMDA) and the National Institute of Biomedical Innovation (NIBI). We thank Ms. Mika Higashida and Ms. Hisae Shiina, and Dr. Kei Fujimoto and Dr. Kanae Yasuda for their help in data and sample collection.
Author information
Authors and Affiliations
Corresponding author
Additional information
F. Takeuchi, Y. Ochiai and M. Serizawa have contributed equally to this work.
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Takeuchi, F., Ochiai, Y., Serizawa, M. et al. Search for type 2 diabetes susceptibility genes on chromosomes 1q, 3q and 12q. J Hum Genet 53, 314–324 (2008). https://doi.org/10.1007/s10038-008-0254-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-008-0254-6
Keywords
This article is cited by
-
Oligopeptide/Histidine Transporter PHT1 and PHT2 — Function, Regulation, and Pathophysiological Implications Specifically in Immunoregulation
Pharmaceutical Research (2023)
-
Dectin-2 mediates phagocytosis of Lactobacillus paracasei KW3110 and IL-10 production by macrophages
Scientific Reports (2021)
-
Regulation and biological role of the peptide/histidine transporter SLC15A3 in Toll-like receptor-mediated inflammatory responses in macrophage
Cell Death & Disease (2018)
-
Di- and tripeptide transport in vertebrates: the contribution of teleost fish models
Journal of Comparative Physiology B (2017)
-
Quantitative assessment of the variation in IGF2BP2 gene and type 2 diabetes risk
Acta Diabetologica (2012)
