
Abstract
Hereditary hearing impairment (HI) displays extensive genetic heterogeneity. To date, 67 autosomal recessive nonsyndromic hearing impairment (ARNSHI) loci have been mapped, and 24 genes have been identified. This report describes three large consanguineous ARNSHI Pakistani families, all of which display linkage to marker loci located in the genetic interval of DFNB49 locus on chromosome 5q13. Recently, Riazuddin et al. (Am J Hum Genet 2006; 79:1040–1051) reported that variants within the TRIC gene, which encodes tricellulin, are responsible for HI due to DFNB49. TRIC gene sequencing in these three families led to the identification of a novel mutation (IVS4 + 1G > A) in one family and the discovery of a previously described mutation (IVS4 + 2T > C) in two families. It is estimated that 1.06% (95% confidence interval 0.02–3.06%) of families with ARNSHI in Pakistan manifest HI due to mutations in the TRIC gene.
Similar content being viewed by others


Introduction
Due to the complexity of the inner ear, alterations in different pathways can lead to the hearing impairment (HI) phenotype. As a consequence, HI etiology exhibits extreme genetic heterogeneity. The prevalence of HI due to genetic factors has been estimated to be at least 1 in 2,000 individuals (Mehl and Thomson 1998, 2002). For nonsyndromic HI (NSHI), autosomal recessive NSHI (ARNSHI) is the most common hereditary form, accounting for >70% of cases (Morton 1991). ARNSHI is usually due to a sensorineural defect (Petit 1996) and is generally prelingual, not progressive, and all frequencies are affected, resulting in severe to profound HI. To date, 67 loci have been mapped for ARNSHI, and 24 genes have been identified (http://webhost.ua.ac.be/hhh/). The clinical usefulness of knowing hundreds of HI genes will remain limited until individual genes are studied for their spectrum of sequence variants, their mutation prevalence rates in various populations are known, and their functional significance in the auditory system is elucidated. Recently, Riazuddin et al. (2006) reported on mutations in the tricellulin encoding the TRIC gene in eight families with ARNSHI due to the DFNB49 locus, which maps to chromosome 5q13 (Ramzan et al. 2005; Riazuddin et al. 2006). Tricellulin is a tricellular tight-junction (tTJ) protein, which is key in the formation of barriers between tricellular contacts of epithelial cells throughout the body (Ikenouchi et al. 2005).
The study presented here describes the identification of a novel homozygous splice-site mutation (IVS4 + 1G > A) in the TRIC gene identified in consanguineous Pakistani family 4,348. Additionally, for two of the consanguineous Pakistani families (4,129 and 4,252), a previously described mutation (IVS4 + 2T > C) in the TRIC gene was found. All three families have profound ARNSHI and displayed linkage to markers located within the DFNB49 genetic interval on chromosome 5q13.
Materials and methods
Ascertainment of families
Approval of the study was obtained from the Institutional Review Boards of Quaid-i-Azam University, Islamabad, Pakistan, and Baylor College of Medicine, TX, USA. Signed informed consent was obtained from all participating subjects. Once a family with ARNSHI is ascertained, two hearing impaired family members are screened for mutation in the GJB2 gene using methods that have previously been described (Santos et al. 2005). Families negative for GJB2 functional variants undergo an genome screening using either ∼400 microsatellite or ∼6,000 single nucleotide polymorphism (SNP) marker loci. To date, 284 Pakistani families with ARNSHI have been ascertained with at least two family members presenting with prelingual HI. Approximately 95% of the hearing impaired individuals are offspring of consanguineous matings. Of the families ascertained with ARNSHI, eight pedigrees have family members who are hearing impaired due to mutations in the GJB2 gene, and an additional 276 families have undergone a whole genome scan. Of those families in which a whole genome scan was produced, three consanguineous families—pedigree 4,129, 4,252, and 4,348 (Fig. 1)—displayed evidence of linkage to the DFNB49 locus, for which mutations in the TRIC gene are known to be responsible for ARNSHI (Riazuddin et al. 2006).

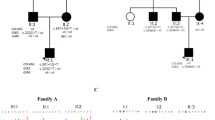
Pedigree drawings for three Pakistani families (4,129, 4,252, and 4,348) with autosomal recessive non-syndromic hearing loss due to mutations in the TRIC gene. Filled symbols represent hearing-impaired individuals. Clear symbols represent unaffected individuals. Below pedigree drawings, audiograms are displayed for hearing-impaired individuals VI-1 (pedigree 4,129), IV-2 (pedigree 4,252), and III-2 (pedigree 4,348) that demonstrate profound hearing impairment, which involves all frequencies in both ears. Audiograms for the normal family members, 4,129 (VI-9), 4,252 (III-2), and 4,348 (II-3), are also displayed for comparison. Circles and crosses represent air conduction for right and left ear, respectively
Two of these families (4,129 and 4,252) originate from the Saraiki-speaking area in Punjab province, and the third family (4,348) originates from the Pashto-speaking area in the North-West Frontier province. An audiometric evaluation was performed for all affected individuals by measuring the threshold of hearing at 250–8,000 Hz for pure-tone air conduction. All affected individuals of the three families (4,129, 4,252, and 4,348) displayed congenital, bilateral, prelingual, profound HI that affects all frequencies (Fig. 1). Features of visual loss, limb deformity, mental deficiency, or gross vestibular involvement were not observed in any affected individual.
Genotyping
Three different approaches were used to map the gene underlying ARNSHI in families 4,129, 4,252, and 4,348. For family 4,348, homozygosity mapping was carried out by genotyping all four affected individuals for 52 microsatellite markers within regions containing genes known to be involved in ARNSHI. All four family members were homozygous for three markers in the region of the DFNB49 locus. All pedigree members for whom DNA was available were genotyped for ten additional microsatellite markers within the region. For families 4,129 and 4,252, whole genome scans were carried out at the National Heart, Lung and Blood Institute (NHLBI) Mammalian Genotyping Service (Marshfield, WI, USA) and the Center for Inherited Disease Research (CIDR) (Bethesda, MD, USA), respectively, using all available informative pedigree members. For family 4,129, the whole genome scan was carried out using 399 fluorescently labeled short tandem repeat markers genotyped across 22 autosomes and X and Y chromosomes spaced approximately every 10 cM. An additional 19 fine-mapping marker loci were genotyped in the region of DFNB49 upon completion of the whole genome scan analysis. For family 4,252 the whole genome scan was carried out using the Illumina panel IV that contains 5,858 SNP marker loci.
Linkage analysis
PEDCHECK (O’Connell and Weeks 1998) was used to verify the absence of Mendelian inconsistencies. Two-point linkage analysis was carried out using the MLINK program of the FASTLINK computer package (Cottingham et al. 1993), and multipoint linkage analysis was performed using ALLEGRO (Gudbjartsson et al. 2005). The genetic distances were determined using the Rutgers Combined Linkage–Physical Map of the Human Genome (build 36) (Kong et al. 2004). For the analysis, an autosomal recessive completely penetrant mode of inheritance was used with a disease allele frequency of 0.001. For families 4,129 and 4,252, marker allele frequencies were estimated using founders and reconstructed founders from 35 and 77 Pakistani pedigrees, respectively, which underwent a genome scan at the same time. For families 4,129 and 4,348, equal allele frequencies were used for the fine-mapping markers because it was not possible to estimate allele frequencies from the founders, as these markers were only genotyped in these families.
Mutational analysis
To screen for the mutation in the TRIC gene, all of its seven exons and splice junctions were polymerase chain reaction (PCR)-amplified from genomic DNA by seven sets of primers designed using Primers3 software (Rozen and Skaletsky 2000). The amplified products were sequenced in an ABI prism 310 Automated DNA Sequencer using the ABI Big Dye Terminator v3.1 cycle sequencing kit (PE Applied Biosystem, Foster City, CA, USA) following purification by Rapid PCR Purification System (Marligen Biosciences, Ijamsville, MD, USA). Sequence data were compared with published sequence of TRIC using the BioEdit Sequence Alignment Editor v7.5.0.2 software package (Hall 1999). The mutation position was assigned according to mRNA sequence from GenBank entry NM_001038603.1, with nt 1 as first nucleotide of the start codon.
Results
For family 4,129, a maximum two-point logarithm of odds (LOD) score of 4.0 (θ = 0.0) and multipoint LOD score of 5.3 was obtained at marker D5S2041. The region of homozygosity for family 4129 spanned from position 58,712,926 to 76,757,809 on chromosome 5 and covered 18.0 Mb according to build 36. For family 4,252, a maximum multipoint LOD score of 4.3 was obtained for seven SNP (rs1168402, rs1961760, rs1200485, rs283596, rs255592, rs586853, and rs1993370) marker loci. The region of homozygosity observed for pedigree 4,252 spanned on chromosome 5 from base 66,424,155 to 78,873,312 and contained 12.2 Mb. For family 4,348, only markers (D5S2003, D5S647, D5S2107) in the region of DFNB49 were homozygous in all four affected family members. The fine mapping of markers within the region yielded a maximum 2-point LOD score of 1.8 with markers D5S2107, D5S2500, D5S2089, D5S647, D5S629, and D5S2003 and maximum multipoint LOD score of 2.4 with markers D5S407, D5S2102, D5S2107, and D5S2500. It should be noted that this family structure does not have a sufficient number of informative meioses to establish linkage. For family 4,348, the region of homozygosity was 16.4 Mb and spanned on chromosome 5 from position 58,185,731 to 74,563,579 on chromosome 5 according to build 36.
Sequencing TRIC
Riazuddin et al. (2006) reported the identification of functional sequence variants in the TRIC gene in eight Pakistani families with ARNSHI mapped to the DFNB49 locus. Therefore, the coding region and splice junction sites of the same gene were sequenced in affected and normal individuals in the families investigated here. Sequence analysis of exon 4 of the gene from nine affected individuals (V-3, VI-1, VI-2, VI-3, VI-4, VI-5, V-6, VI-7, VI-8) in family 4,129 and four affected individuals (IV-2, IV-3, IV-4, IV-5) in family 4,252 revealed a T to C transition at splice-donor site of intron 4 (IVS4 + 2T > C). In family 4,348, sequence analysis of the same exon 4 of the gene from four affected individuals (III-2, III-3, III-4, III-5) identified a G to A transition at splice-donor site of intron 4 (IVS4 + 1G > A). These two splice-site mutations were present in the heterozygous state in obligate carriers within the families (Fig. 2). To ensure that the mutations did not represent benign polymorphisms within the Pakistani population, a panel of 100 unrelated unaffected individuals (200 chromosomes) was screened for the two mutations, and these variants were not observed in control panel DNA samples.

Automated DNA sequence electropherograms of TRIC-gene mutations. Sequence analysis of exon 4 in normal (a), obligate carrier (b), and affected (c) individuals of families 4,129 and 4,252. The recurrent splice-site mutation (IVS4 + 2T > C) was identified in families 4,129 and 4,252. Sequence analysis of exon 4 in normal (d), obligate carrier (e), and affected (f) individuals of family 4,348. The splice-site mutation (IVS4 + 1G > A) was found in family 4,348
Based upon the sample of ARNSHI families ascertained from Pakistan, it is estimated that the prevalence of ARNSHI due to mutations in the TRIC gene is 1.06% (95% confidence interval 0.02–3.06%)
Discussion
In this study, we identified a novel splice-site mutation (IVS4 + 1G > A) and a previously observed mutation (IVS4 + 2T > C) in the TRIC gene in three Pakistani consanguineous families, which were linked to marker loci in the DFNB49 genetic interval on chromosome 5q13. Both mutations alter the splice-donor site of intron 4 of the TRIC gene and are predicted to affect mRNA splicing. Previously, Riazuddin et al. (2006) reported two mutations in the splice-donor site of exon 4 (IVS4 + 2T > C and IVS4 + 2delTGAG), a mutation in the splice-acceptor site of exon 4 (IVS3 − 1G > A), and a transition mutation in exon 5 (c.1498C > T) of the TRIC gene in the families, which showed linkage to the DFNB49 locus. Families 4,129 and 4,252, which presented with the IVS4 + 2T > C mutation, belong to the Punjabi-speaking population in the Punjab province of Pakistan. The origin of the four families with the same mutation reported by Riazuddin et al. (2006) is unknown. These four families all displayed the same haplotype, thus strongly suggesting that the mutation observed in these families was due to a single mutation event. For families 4,129 and 4,252, it was not possible to elucidate whether or not they share the same haplotype as the families reported by Riazuddin et al. (2006) or share a different haplotype. DNA samples from family 4,252 were only genotyped for SNP marker loci, and although the DNA samples from family 4,129 were genotyped using microsatellite marker loci, none of the marker loci within the region of homozygosity overlapped with those reported by Riazuddin et al. (2006).
In the inner-ear epithelia, tricellulin is concentrated in tTJs (Riazuddin et al. 2006), where the fluid-filled compartments of different ionic compositions of the organ of Corti are required for normal hearing (Kitajiri et al. 2004). Riazuddin et al. (2006) demonstrated the presence of at least four alternatively spliced TRIC isoforms (TRIC-a, TRIC-a1, TRIC-b, TRIC-c) in various mouse tissues. The longest TRIC mRNA (TRIC-a), detected in human fetal cochlear mRNA, has seven exons (GenBank entry NM_001038603.1) and is predicted to encode 558 amino acids containing four transmembrane domains encoded by exon 2 and 103 amino acid occludin-ELL domain (Pfam accession number PF07303) encoded by exons 5, 6, and 7 of the TRIC gene. The TRIC-a1 isoform (GenBank accession number DQ682657) lacks exon 3 but contains an occludin-ELL domain. The COOH-terminal sequence (∼130 aa) is conserved between tricellulin and occludin; in occludin, this region was reported to be remarkably conserved among various vertebrate species (Ando-Akatsuka et al. 1996) and to contain a domain responsible for its association with scaffolding protein ZO-1, a major TJ plaque protein (Furuse et al. 1993).
The three exon 4 splice sites and a nonsense mutation in the TRIC gene identified in eight families studied by Riazuddin et al. (2006) and splice-site mutations identified in the study reported here are predicted to generate truncated proteins that are unable to interact with the three scaffolding proteins ZO-1, ZO-2, and ZO-3. Full-length TRIC is thought to be essential for the high tightness of the TJs of the organ of Corti (Riazuddin et al. 2006). Thus, it is conceivable that the mutations in the TRIC gene that affect exon 4 could lead to truncated TRIC protein in the inner ear epithelias, which is then unable to produce effective TJs for hearing.
References
Ando-Akatsuka Y, Saitou M, Hirase T, Kishi M, Sakakibara A, Itoh M, Yonemura S, Furuse M, Tsukita S (1996) Interspecies diversity of the occludin sequence: cDNA cloning of human, mouse, dog, and rat-kangaroo homologues. J Cell Biol 133:43–47
Cottingham R, Indury RM, Schaffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263
Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S (1993) Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 123:1777–1788
Gudbjartsson DF, Thorvaldsson T, Kong A, Gunnarsson G, Ingolfsdottir A (2005) Allegro version 2. Nat Genet 37:1015–1016
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp 41:95–98
Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, Tsukita S (2005) Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol 171:939–945
Kitajiri S, Miyamoto T, Mineharu A, Sonoda N, Furuse K, Hata M, Sasaki H, Mori Y, Kubota T, Ito J, Furuse M, Tsukita S (2004) Compartmentalization established by claudin-11-based tight junctions in stria vascularis is required for hearing through generation of endocochlear potential. J Cell Sci 117:5087–5096
Kong X, Murphy K, Raj T, He C, White PS, Matise TC (2004) A combined linkage-physical map of the human genome. Am J Hum Genet 75:1143–1148
Mehl AL, Thomson V (1998) Newborn hearing screening: the great omission. Pediatrics 101:e4
Mehl AL, Thomson V (2002) The Colorado newborn hearing screening project, 1992–1999: on the threshold of effective population-based universal newborn hearing screening. Pediatrics 109:e7
Morton NE (1991) Genetic epidemiology of hearing impairment. Ann NY Acad Sci 630:16–31
O’Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259–266
Petit C (1996) Genes responsible for human hereditary deafness: symphony of a thousand. Nat Genet 14:385–391
Ramzan K, Shaikh RS, Ahmad J, Khan SN, Riazuddin S, Ahmed ZM, Friedman TB, Wilcox ER, Riazuddin S (2005) A new locus for nonsyndromic deafness DFNB49 maps to chromosome 5q12.3-q14.1. Hum Genet 116:17–22
Riazuddin S, Ahmed ZM, Fanning AS, Lagziel A, Kitajiri S, Ramzan K, Khan SN, Chattaraj P, Friedman PL, Anderson JM, Belyantseva IA, Forge A, Riazuddin S, Friedman TB (2006) Tricellulin is a tight-junction protein necessary for hearing. Am J Hum Genet 79:1040–1051
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Santos RL, Wajid M, Pham TL, Hussan J, Ali G, Ahmad W, Leal SM (2005) Low prevalence of Connexin 26 (GJB2) variants in Pakistani families with autosomal recessive non-syndromic hearing impairment. Clin Genet 67:61–68
Acknowledgments
We thank the patients and other family members for participating in this study. The work presented here was funded by the Higher Education Commission (HEC), Islamabad, Pakistan, and National Institutes of Health (NIH), National Institute of Deafness and other Communication Disorders (NIDCD), Bethesda, MD, USA, grant DC03594. Muhammad Salman Chishti was supported by indigenous Ph.D. fellowships from HEC and NIDCD grant R01-DC03594. Genotyping services were provided by Center for Inherited Disease Research (CIDR) (Bethesda, MD, USA) and the National Heart, Lung and Blood Institute (NHLBI) Mammalian Genotyping Service (Marshfield, WI, USA). CIDR is fully funded through a federal contract from the NIH to The Johns Hopkins University (Baltimore, MD, USA), contract number N01-HG-65403.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chishti, M.S., Bhatti, A., Tamim, S. et al. Splice-site mutations in the TRIC gene underlie autosomal recessive nonsyndromic hearing impairment in Pakistani families. J Hum Genet 53, 101–105 (2008). https://doi.org/10.1007/s10038-007-0209-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-007-0209-3
