
Abstract
Hearing loss (HL) is one of the most common sensory defects, of which X-linked nonsyndromic hearing loss (NSHL) accounts for only 1–2%. While a COL4A6 variant has been reported in a single Hungarian family with NSHL associated with inner ear malformation, causative role of COL4A6 variants and their phenotypic consequences in NSHL remain elusive. Here we report two families in which we identified a male member with X-linked HL. Each has inherited a rare hemizygous COL4A6 variant from their respective mothers, NM_001287758.1: c.3272 G > C (p.Gly1091Ala) and c.951 + 1 G > C. An in vitro minigene splicing assay revealed that c.951 + 1 G > T leads to skipping of exon 15, strongly suggesting a pathogenic role for this variant in the HL phenotype. The p.Gly1091Ala variant is classified as a variant of unknown significance based on the variant interpretation guidelines. This report provides evidence for variants in the COL4A6 gene resulting in X-linked NSHL. It highlights the importance of in-depth genetic studies in all family members in addition to the proband, especially in multiplex families, to determine the precise etiology of HL.
Similar content being viewed by others


Introduction
One out of every 1000 live births is affected with severe to profound congenital hearing loss (HL), and 50% of the sensorineural HL cases are hereditary in nature. Approximately 80% of nonsyndromic hearing loss (NSHL) cases are inherited in an autosomal recessive manner, while 20% are autosomal dominant and 1–2% are X-linked. Autosomal recessive mutations in GJB2 are by far the most common genetic etiology and account for more than 30% of NSHL in children in Western Europe. From more than 120 deafness genes/loci reported in literature to date, six loci (DFNX1-6) with five genes cloned (PRPS1, POU3F4, SMPX, AIFM1, and COL4A6), have been implicated in X-linked NSHL [1]. Many syndromic X-linked disorders are associated with HL, such as Norrie syndrome, X-linked deafness-dystonia-optic neuropathy, and Alport syndrome presenting with HL as a major clinical finding [1]. Interestingly, Alport syndrome, a heterogeneous condition characterized by nonimmune glomerulopathy and progressive sensorineural HL can be inherited as an autosomal dominant, autosomal recessive, or X-linked condition. The X-linked forms accounts for ~70% of Alport syndrome cases and is associated with variants in the COL4A5 gene [2,3,4]. A contiguous X-chromosome deletion involving COL4A5 and COL4A6 presents with X-linked Alport syndrome with HL and leiomyomatosis in the gastrointestinal, respiratory, and female reproductive tracts [5,6,7]. The autosomal forms of Alport syndrome are less common compared to the X-linked type and occur due to sequence variants in the COL4A3 and COL4A4 genes [8]. The COL4A6 gene on Xq22.3 encodes a classical type IV collagen with a close homology to all the other five chains. Its protein structure most closely resembles that of COL4A2 and COL4A4 [9]. COL4A6 is expressed in the stria vascularis and spiral ganglia cells in the mouse inner ear, and the alpha-6-chain of collagen type IV is a part of the basement membrane of the inner ear and is thought to have an important role in development of the cochlea. Being a member of the collagen family, COL4A6 forms a triple helical structure with two other alpha-5-chains, which is stabilized by the presence of glycine as every third residue. We hypothesize that the presence of a splice variant with skipping of an exon in Family A and a mutation of a glycine residue at the highly conserved residue, Gly1091Ala in Family B can result in destabilization of the collagen molecule with inner ear abnormalities and HL.
Here we present data on two families from North America segregating two separate variants in the COL4A6 gene as a cause of their X-linked NSHL. Both probands had deaf parents and were identified from a larger subgroup of families within a North American Repository of DNA from individuals with HL [10]. DNA from all samples underwent initial GJB2 and GJB6 testing, mitochondrial testing, and subsequent testing for a deafness gene panel using NGS. The COL4A6 variants identified by NGS were confirmed by Sanger sequencing in the proband and family members. Further in silico and splicing analyses reaffirm our hypothesis that the two identified variants in the COL4A6 gene are a plausible basis for their HL.
Material
Patient ascertainment
Deaf probands from North America and their family members were ascertained for this study through several sources, including the Annual Survey of Deaf and Hard of Hearing Children and Youth, conducted at the Gallaudet Research Institute of Gallaudet University (GU), an institution of higher education for the deaf and hard of hearing. The Annual Survey collected educational, etiologic, and audiologic data, as well as demographic information such as race, parental mating type, and hearing status of siblings on a nationwide sample of nearly 50,000 deaf and hard of hearing students who receive special education services because of their HL [10]. Participants were also recruited through the GU Alumni Association [11]. All participants completed the informed consent process approved by the GU Institutional Review Board.
Audiological testing
Pure tone audiometry was performed according to current standards to determine air conduction thresholds at 0.125, 0.25, 0.5, 1, 2, 4, 6, and 8 kHz. Audiological data were available only in certain deaf members from Family B. The audiogram for individual B-II:3 revealed profound bilateral HL.
Clinical history
Proband A-III:1, ascertained through GU Alumni Association, was 24 years old and reported severe HL (Fig. 1A). There was no report of any skin findings, renal abnormalities, or ophthalmologic changes. Although we did not receive audiograms from the family, parents of A-III:1 are also reported to have HL. The father, individual A-II:1, has deaf parents and a deaf brother. The mother, A-II:2, reported HL from high fever during a varicella infection as a child.

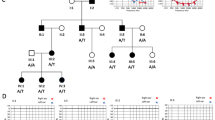
A, B Families A and B with their multiplex pedigrees and segregation for variants in GJB2, GJB6, and COL4A6. C Electropherogram for Sanger sequencing of the hemizygous male (A-III:1 and B-II:3), heterozygous mother (A-II:2 and B-I:2), and unaffected father (A-II:1 and B-I:1).
Proband B-III:1 was ascertained from an audiology clinic in Mexico at 18 months of age. He had congenital deafness, no response on ABR testing, and did not cooperate for a behavioral hearing test. His father, paternal uncle, and mother (B-II:3, B-II:2, B-II:4) are reported to have severe to profound HL with a prelingual onset. The paternal grandmother, B-I:2, reported premature graying and was noted to have dystopia canthorum. No additional clinical or phenotypic details were available.
Cranial or temporal bone imaging studies are not available for Families A and B.
Methods
Molecular testing
DNA from Families A and B was extracted from peripheral blood samples using Pure Gene (Gentra Systems) protocols. DNA samples from these two families were screened for mutations in exons 1 and 2 of GJB2 by Sanger sequencing as described in Pandya et al. [10]. The del (GJB6-D13S1830) deletion was tested using primers described by del Castillo et al. [12]. Mitochondrial testing was performed as described by Pandya et al. [13]. Additional NGS was performed on a panel of 187 HL genes as described in Yan et al. [14]. A microarray or whole-exome sequencing (WES) was not performed.
Minigene splicing assay and sequencing of in vitro transcript
In vitro minigene assay was performed as described by Booth et al. [15]. Briefly, wild-type exons 15 and 16 and ~360 base pairs of each flanking intron of COL4A6 were polymerase chain reaction (PCR) amplified and ligated into the pET01 vector (MoBiTec, Goettingen, Germany). The c.951 + 1 G > T variant was introduced to the wild-type vector according to the manufacturer’s protocol. Wild-type and mutant vectors were transfected into HEK293 cells. Total RNA was harvested 48 h post transfection and complementary DNA (cDNA) was transcribed according to the manufacture protocol. PCR using primers specific to the 5′ and 3′ native exons of the pET01 vector was performed and products were visualized on an agarose gel. Gel products were extracted and Sanger sequenced.
Pathogenicity determination tools
COL4A6 variants identified in the family members were interpreted according to the ACMG guidelines along with its recent modification for hereditary HL [16].
Predictions of pathogenicity for the variants of interest were made was using Adjusted CADD scores [17], MutationT@ster2 [18], PolyPhen-2 [19], and SIFT [20].
Results
Variant discovery
Family A
Initial screening of GJB2, GJB6, and MT-RNR1 in the proband A-III:1 identified the dominant variant in GJB2 c.223 C > T (p.Arg75Trp) that was paternally inherited (Fig. 1A). A heterozygous GJB6 [del (GJB6-D13S1854)] was noted in the mother A-II:2 but not in the proband. Additionally, the HL NGS gene panel identified a maternally inherited donor splice site variant c.951 + 1 G > T in COL4A6 (Fig. 1C). The splice site variant in COL4A6 c.951 + 1 G > T affects the canonical GT dinucleotide consensus sequence at the donor site of intron 14. The splicing assay (Fig. 2) for the COL4A6 c.951 + 1 G > T variant reveals skipping of exon 15 with introduction of a premature stop codon, further confirmed by Sanger sequencing the pET01_COL4A6 cDNA transcript (Fig. 2C). ACMG variant interpretation guidelines and our minigene study suggest the variant is likely pathogenic (Table 1) [21].

A Depiction of the altered splice acceptor site in intron 15 (TT) which is bypassed. B Verification of pET01-COL4A6 transfection. Wild-type and mutant cDNA harvested and transcribed from pET01-COL4A6 transfected HEK293 cells. Agarose gel shows 200 bp wild-type product that includes exons 15 and 16, and a 150 bp product with exon 15 skipped in the mutant c.951 + 1 G > T lane. C Sanger sequence confirmation of extracted product noted in agarose gel in (B).
Family B
The cause for HL in the proband B-III:1 remained uncertain following the initial identification of a heterozygous GJB2 c.35delG variant (Fig. 1B), which is involved in autosomal recessive NSHL. Subsequent testing with the HL gene panel in the trio also failed to identify an etiology in the proband. Subsequently, we identified a COL4A6 variant c.3272 G > C (p.Gly1091Ala) in the father of the proband, B-II:3 that was also present in his mother B-I:2 and brother B-II:2 who both have HL (Fig. 1C). Pathogenicity tools predict that the variant is disease causing (Table 1). It generated a CADD score of 23 and disrupts the Gly-X-Y motif in COL4A6. ACMG variant interpretation guidelines suggest it to be a variant of unknown significance (Table 1) [21].
Discussion
Family A
The GJB2 p.Arg75Trp variant in heterozygous state has been identified in at least 11 individuals with HL [22,23,24,25,26,27]. Most reported individuals had severe to profound HL that was either congenital or progressive in infancy, and some individuals also had palmoplantar keratoderma. This variant occurred de novo in three individuals and was identified in three affected parents, which supports that this variant is inherited in a dominant manner [22, 25,26,27,28]. Additionally the p.Arg75Trp variant has not been identified in large population studies. The functional assay demonstrated that the change to a Tryptophan (Trp) at position 75 has a dominant negative affect on the protein, disrupts the function of the gap junction [21, 28,29,30], and is important for the postnatal development of the organ of Corti and normal hearing [31, 32].
The novel splice site variant in COL4A6 c.951 + 1 G > T is not reported in the ClinVar database and has a minor allele frequency of 0.00001 (1/177373, gnomAD). The inappropriately spliced mRNA with the loss of exon 15 results in a disrupting 15-amino-acid deletion and aberrant protein suggesting it as a clinically significant alteration.
In the proband, we hypothesize that both the COL4A6 and GJB2 pathogenic variants contribute to the HL and postulate the HL to be more severe than in either of his parents; however, we did not obtain audiometric data to confirm this hypothesis. The father’s HL is attributed to the dominant GJB2 variant, and although we do not have audiometric data for the mother, heterozygous carriers for the COL4A6 gene have been reported to exhibit mild to moderate HL [33]. The contribution of the GJB6 [del (GJB6-D13S1854)] deletion, an autosomal recessive variant, to HL in the mother is not likely to be significant. Trying to identify an etiology in the proband that also explains the HL in the parents in this family contributed to defining a more precise molecular etiology.
Family B
The COL4A6 variant c.3272 G > C (p.Gly1091Ala) identified in the proband’s father has an allele frequency of 0.001075 in the Latino population (gnomAD). Based on the segregation in the family with deaf members, pathogenicity tool prediction, and disruption of the Gly-X-Y motif in the COL4A6 protein, we propose this is the cause of deafness in B-II:2 and his brother B-II:3 (variant call using SVI), although the etiology in the proband remains to be determined. It certainly helps clarify that the father will not pass this variant to his sons but that his daughters would be carriers.
Conclusions
This report adds highly relevant data to the previously reported genes implicated in nonsyndromic X-linked sensorineural HL. To date, only one other family with a COL4A6 sequence variant has been reported with HL in which the index proband and his mother were identified with a missense mutation in exon 23 of the COL4A6 gene, c.1771 G > A p.Gly591Ser, which segregated in other affected male members in the family with HL [33]. The present report confirms the role of COL4A6 variants in in two additional families with X-linked NSHL.
Although the dominant GJB2 variant explained HL in Family A, a family structure with two deaf parents persuaded us to look further. While WES data were not available in these cases and could have provided additional insight, the comprehensive NGS panel resulted in the identification of the novel COL4A6 likely pathogenic variant in the proband. This finding has genetic counseling implications given the different inheritance pattern for each variant. Similarly, in Family B we are yet to identify an etiology for the HL in the proband, however, the dense family history of HL made us probe further in the older generations yielding the COL4A6 variant in the proband’s father as a likely explanation of his and his brothers HL. This information allows more precise genetic counseling for Family A due to the proband’s offspring having a 50% risk of developing HL due to the GJB2 variant, and all his daughters (and none of his sons) inheriting the COL4A6 variant with variable expression that can be passed on to his grandsons. Thus, caution needs to be exercised when evaluating deaf individuals when both parents have HL. When determining the etiology of HL, clinicians should strive to utilize current technology to having a precise molecular etiology as it enables more accurate counseling for recurrence risk assessment and prognosis.
References
Corvino V, Apisa P, Malesci R, Laria C, Auletta G, Franze A. X-linked sensorineural hearing loss: a literature review. Curr Genomics. 2018;19:327–38.
Knebelmann B, Breillat C, Forestier L, Arrondel C, Jacassier D, Giatras I, et al. Spectrum of mutations in the COL4A5 collagen gene in X-linked Alport syndrome. Am J Hum Genet. 1996;59:1221.
Plant KE, Green PM, Vetrie D, Flinter FA. Detection of mutations in COL4A5 in patients with Alport syndrome. Hum Mutat. 1999;13:124–32.
Wang F, Ding J, Yu Lx, Yang Jy. Detection of COL4A5 gene mutations in Alport syndrome by analyzing cDNA of skin fibroblasts. J Beijing Med Univ. 2002;3:219–224.
Mothes H, Heidet L, Arrondel C, Richter KK, Thiele M, Patzer L, et al. Alport syndrome associated with diffuse leiomyomatosis: COL4A5‐COL4A6 deletion associated with a mild form of Alport nephropathy. Nephrol Dial Transplant. 2002;17:70–4.
Uliana V, Marcocci E, Mucciolo M, Meloni I, Izzi C, Manno C, et al. Alport syndrome and leiomyomatosis: the first deletion extending beyond COL4A6 intron 2. Pediatr Nephrol. 2011;26:717–24.
Sá MJN, Fieremans N, de Brouwer AP, Sousa R, e Costa FT, Brito MJ, et al. Deletion of the 5′ exons of COL4A6 is not needed for the development of diffuse leiomyomatosis in patients with Alport syndrome. J Med Genet. 2013;50:745–53.
Kashtan CE. Alport Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021.
Zhou J, Ding M, Zhao Z, Reeders ST. Complete primary structure of the sixth chain of human basement membrane collagen, alpha 6(IV). Isolation of the cDNAs for alpha 6(IV) and comparison with five other type IV collagen chains. J Biol Chem. 1994;269:13193–9.
Pandya A, Arnos KS, Xia XJ, Welch KO, Blanton SH, Friedman TB, et al. Frequency and distribution of GJB2 (connexin 26) and GJB6 (connexin 30) mutations in a large North American repository of deaf probands. Genet Med. 2003;5:295–303.
Arnos KS, Welch KO, Tekin M, Norris VW, Blanton SH, Pandya A, et al. A comparative analysis of the genetic epidemiology of deafness in the United States in two sets of pedigrees collected more than a century apart. Am J Hum Genet. 2008;83:200–7.
del Castillo FJ, del Castillo I, Moreno-Pelayo MA, Brownstein Z, Marlin S, Adina Q, et al. Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: a multicenter study. Am J Hum Genet. 2003;73:1452–8.
Pandya A, Xia X, Erdenetungalag R, Amendola M, Landa B, Radnaabazar J, et al. Heterogenous point mutations in the mitochondrial tRNA Ser (UCN) precursor coexisting with the A1555G mutation in deaf students from Mongolia. Am J Hum Genet. 1999;65:1803–6.
Yan D, Tekin D, Bademci G, Foster J, Cengiz FB, Kannan-Sundhari A, et al. Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Hum Genet. 2016;135:953–61.
Booth KT, Azaiez H, Kahrizi K, Wang D, Zhang Y, Frees K, et al. Exonic mutations and exon skipping: lessons learned from DFNA5. Hum Mutat. 2018;39:433–40.
Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat. 2018;39:1593–613.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248.
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
Richard G, White TW, Smith LE, Bailey RA, Compton JG, Paul DL, et al. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet. 1998;103:393–9.
Putcha GV, Bejjani BA, Bleoo S, Booker JK, Carey JC, Carson N, et al. A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genet Med. 2007;9:413.
Mani RS, Ganapathy A, Jalvi R, Srisailapathy CS, Malhotra V, Chadha S, et al. Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur J Hum Genet. 2009;17:502.
Yuan Y, Huang D, Yu F, Zhu X, Kang D, Yuan H, et al. A de novo GJB2 (connexin 26) mutation, R75W, in a Chinese pedigree with hearing loss and palmoplantar keratoderma. Am J Med Genet Part A. 2009;149:689–92.
Lee JY, In S, Kim HJ, Jeong S, Choung YH, Kim YC. Hereditary palmoplantar keratoderma and deafness resulting from genetic mutation of Connexin 26. J Korean Med Sci. 2010;25:1539–42.
Weegerink NJ, Pennings RJ, Huygen PL, Hoefsloot LH, Cremers W, Kunst HP. Phenotypes of two Dutch DFNA3 families with mutations in GJB2. Ann Otol Rhinol Laryngol. 2011;120:191–7.
Pang X, Chai Y, Sun L, Chen D, Chen Y, Zhang Z, et al. Characterization of spectrum, de novo rate and genotype-phenotype correlation of dominant GJB2 mutations in Chinese hans. PLoS ONE. 2014;9:e100483.
Deng Y, Chen Y, Reuss L, Altenberg GA. Mutations of connexin 26 at position 75 and dominant deafness: essential role of arginine for the generation of functional gap-junctional channels. Hear Res. 2006;220:87–94.
Marziano NK, Casalotti SO, Portelli AE, Becker DL, Forge A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum Mol Genet. 2003;12:805–12.
Inoshita A, Iizuka T, Okamura H, Minekawa A, Kojima K, Furukawa M, et al. Postnatal development of the organ of Corti in dominant-negative Gjb2 transgenic mice. Neuroscience 2008;156:1039–47.
Inoshita A, Karasawa K, Funakubo M, Miwa A, Ikeda K, Kamiya K. Dominant negative connexin26 mutation R75W causing severe hearing loss influences normal programmed cell death in postnatal organ of Corti. BMC Genet. 2014;15:1.
Rost S, Bach E, Neuner C, Nanda I, Dysek S, Bittner RE, et al. Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6. Eur J Hum Genet. 2014;22:208.
Acknowledgements
This study was supported by NIH Grant R01DC009645 from the National Institutes of Health/National Institute on Deafness and Other Communication Disorders.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
O’Brien, A., Aw, W.Y., Tee, H.Y. et al. Confirmation of COL4A6 variants in X-linked nonsyndromic hearing loss and its clinical implications. Eur J Hum Genet 30, 7–12 (2022). https://doi.org/10.1038/s41431-021-00881-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00881-2
This article is cited by
-
New year, new issue
European Journal of Human Genetics (2022)
-
Deafness—family matters
European Journal of Human Genetics (2022)
