
Abstract
Hereditary lymphedema type I (HL-I), also known as Milroy disease, is an autosomal dominant disorder characterized by typical phenotype of infantile onset lower-limb lymphedema accompanied by variable expression of recurrent episodes of cellulites, toenail changes, and papillomatosis. Mutations in the vascular endothelial growth factor receptor 3 (VEGFR3), also known as FLT4 gene, which encodes a lymphatic endothelial-specific tyrosine kinase receptor, have been identified as a genetic cause of HL-I. We report a large Muslim Arab family residing in northern Israel with 14 individuals presenting clinical features of HL-I. Genetic analysis revealed novel missense mutation E1106K in the tyrosine kinase domain II of VEGFR3 that cosegregates with the disorder in the family. Most affected individuals presented with bilateral congenital lower-limb lymphedema. Wide intrafamilial phenotypic variability included two asymptomatic individuals, a case of prenatal hydrothorax evolving to hydrops fetalis, and a late-onset complication, yet unreported, of chronic degenerative joint disease of the knees. This report broadens the known “classic” phenotype of HL-I.
Similar content being viewed by others

Introduction
Primary lymphedema is a disorder caused by dysfunction of the lymphatic system characterized by peripheral edema mainly of the lower extremities. Familial lymphedema is usually segregating as an autosomal dominant disorder and is classified according to the age of onset. Early onset (within the first 2 years of life) is termed hereditary lymphedema type I (HL-I), or Nonne-Milroy disease (MIM 153100). Late onset usually begins after puberty and is termed hereditary lymphedema type II (HL-II) or Meige lymphedema (MIM 153200) (Dale 1985). Lymphangiography often demonstrates lymphatic truncal aplasia or hypoplasia with minimal or no dermal collateral formation (Greenlee et al. 1993). Linkage to chromosomal locus 5q35.3 was reported in some families with HL-I in 1998 and 1999 (Ferrell et al. 1998; Evans et al. 1999). Subsequently, the vascular endothelial growth factor receptor 3 gene (VEGFR3), also known as FLT4, located within this region was found to be the causative gene (Irrthum et al. 2000; Karkkainen et al. 2000). Since then, several mutations were identified. Most of them were missense mutations within the tyrosine kinase domains I or II of the receptor, leading to malfunctioning of the VEGFR3 gene (Irrthum et al. 2000; Karkkainen et al. 2000; Evans et al. 2003). Since VEGFR3 is expressed early in embryogenesis in the lymphatic endothelium, disruption of its activity site (i.e. tyrosine kinase signaling) seems to result in congenital lymphedema (Kim and Dumont 2003). In the present study, we report a large, three-generation family with 16 individuals carrying a novel missense mutation, E1106K, in the tyrosine kinase domain II of the VEGFR3 gene. Fourteen were phenotypically affected and displayed a wide range of clinical features.
Patients and methods
Subjects
The family members are of Muslim Arab origin residing in a village in the northern region of Israel. First, DNA was sampled from two patients with clinical phenotype of HL-I after obtaining an appropriate signed informed consent for VEGFR3 mutation analysis. Following identification of a novel missense mutation in VEGFR3 and the approval of the Institutional Ethical Committee, a pedigree was constructed. and family members, affected and unaffected, were interviewed and clinically examined by two of the authors (RS and SAS). DNA samples were then collected after obtaining signed informed consent from a total of 30 family members, of whom 14 were clinically affected, for genetic analysis of the novel familial VEGFR3 mutation.
Genetic analysis
GeneBank accession number X68203 was used as reference sequence. Exons 16–27 of the VEGFR3 gene encoding for the intracellular tyrosine kinase domain of the receptor were amplified from genomic DNA. Screening was performed using single-strand conformation polymorphism (SSCP) and heteroduplex analysis (HA), followed by direct sequencing, as previously described (Irrthum et al. 2000). Exon 24 of VEGFR3 was amplified using the forward primer GCCCGGCTGCCCCTGAAG and reverse primer GTAACCTGCCGCCAGTGACC. Polymerase chain reaction (PCR) amplification was performed in the presence of betaine 1 M, 66°C annealing temperature, and 35 cycles, resulting in amplification of 171-bp fragment. Cosegregation, as well as screening of 110 unaffected controls, was carried out by restriction fragment length assay. The mutation revealed an additional recognition site for ASP700 endonuclease. The enzyme does not digest the 171-bp wild-type PCR amplicon but cuts once the mutated fragment with c.3316G > A substitution into 100 and 71 bp.
Results
DNA analysis
Results of the DNA analysis revealed a novel heterozygous missense mutation c.3316G > A in the VEGFR3 gene, causing an amino acid substitution p.E1106K in the tyrosine kinase domain II of the receptor. The mutation was found to cosegregate with lymphedema in the family and was present in all the 14 affected members and also in two asymptomatic individuals: II-6 and III-11. Based on the pedigree, II-6 should be an obligate carrier (Fig. 1). The mutation was not present in 110 normal control individuals. Alignment studies showed a high evolutionary conservation for glutamic acid in position 1106 of VEGFR3 amino acid sequence.

Pedigree of the family
Phenotype and pedigree
Figure 1 displays the pedigree of the family. An autosomal dominant pattern of inheritance is evident. Fourteen individuals (seven females and seven males, ranging from 1 month to 68 years of age) exhibited clinical manifestations of HL-I. In all the affected individuals, symptoms were reported to appear within the first year of life; in most of them, the typical features were present since birth. Table 1 summarizes the clinical features of all 16 individuals in the family who were tested and found to carry the VEGFR3 E1106K mutation.
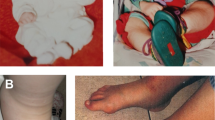
Fourteen individuals (88%) had lower-limb edema. Edema level ranged from the ankle level (six patients) (Fig. 2a) to the knee level (eight patients). Severity of the edema was determined clinically by the examiners and ranged from mild to severe. Seven patients (50%) experienced severe edema, five had moderate edema, and two had mild edema. In 11 affected individuals (78%), edema involved both legs though in some, the involvement was asymmetrical. Lower-limb nail changes (curved nails, Fig. 2b) were present in 13 (92%) of the affected individuals. Four of the seven phenotypically affected males had some degree of hydrocele. Three patients (21%) had papillomatosis of the feet (Fig. 2b). None of the affected patients reported previous episodes of lower-limb cellulites. Five patients (36%) (all older than 40 years) suffered from knee osteoarthritis, a degenerative disease of the joint that necessitated surgical involvement (arthroscopy) in all of them. There was no report of other elderly individuals in this family suffering from knee osteoarthritis. Prenatal severe presentation of in utero bilateral hydrothorax and hydrops fetalis, which necessitated in utero pleural drainage, occurred in one patient (III-22) and resolved spontaneously after birth (Daniel-Spiegel et al. 2005).

a Marked lymphedema with papillomatosis and nail changes in patient II-9. b Congenital bilateral feet lymphedema with up slanting toenails in patient III-22
Discussion
In this study, we identified a novel missense mutation in the VEGFR3 gene, which causes substitution of an evolutionary conserved glutamic acid into lysine in the tyrosine kinase domain II of the receptor. All affected individuals in the family who were tested carried the mutation. Previous studies have shown that mutations affecting either of the two tyrosine kinase domains of VEGFR3 disrupt its autophosphorylation capacity (Irrthum et al. 2000; Karkkainen et al. 2000).
During early development, VEGFR3 is ubiquitously expressed in almost all endothelial cells. However, as development progresses VEGFR3 expression becomes restricted mostly to lymphatic endothelial cells. These findings suggest that the receptor has a critical role in mediating lymphangiogenic proliferation (Kim and Dumont 2003). Abnormal VEGFR3 signaling during embryonic development results in abnormal lymphatic channel network. All VEGFR3 mutations reported so far, including the present one, are missense mutations in either of the two tyrosine kinase domains of the gene, except a single amino acid deletion located within the second tyrosine kinase domain (del F1108) (Evans et al. 2003). Interestingly, most mutations reported to result in HL-I, including ours, were “private” familial mutations (Irrthum et al. 2000; Evans et al. 2003; Brice et al. 2005).
The penetrance of HL-I in our family was 88% and was similar to previous reports (Evans et al. 1999, 2003; Brice et al. 2005). However, analyzing penetrance according to gender, penetrance in females is 100% and in males only 78%. Similar gender differences in penetrance was reported previously by Dale (1985) and Evans et al. (1999) and was attributed to possible different hormonal influence on lymphatic drainage between males and females. Of note, other studies demonstrated similar penetrance in males and females (Witte et al. 1998; Levinson et al. 2003; Evans et al. 2003; Brice et al. 2005); therefore, further studies that include large numbers of affected individuals are needed to better resolve this issue. Factors that influence incomplete penetrance in HL-I are yet to be discovered, but finding in this report that both individuals who carry the mutation and were clinically unaffected are a father (II-6) and his son (III-11), suggesting that other genetic factors (such as modifier genes) may play a significant role.
HL-I is considered to be a well-defined clinical entity with mainly cosmetic consequences of lower limb edema. This study broadens the clinical spectrum of HL-I to include prenatal presentation as well as late onset manifestation. Knee osteoarthritis was found in five individuals (four bilateral, one unilateral), all of whom were older than 40 years and had edema up to the knee level. Joint disease was severe enough to necessitate surgical intervention (arthroscopy). Osteoarthritis is a heterogeneous group of disorders characterized pathologically by focal areas of loss of articular cartilage in synovial joints associated with varying degrees of osteophyte formation, subchondral bone change, and synovitis. Several risk factors are known to predispose to the development of osteoarthritis, including advanced age, obesity, and genetic factors (Dieppe and Lohmander 2005). We hypothesize that the pathogenesis of knee osteoarthritis in our patients may be attributed to local mechanical factors that result from the long-standing lower-limb edema. Chronic degenerative disease of the knees has a major contribution on the morbidity of HL-I. It may be prevented by measures such as manual lymphatic drainage and lower-limb bandaging, avoidance of excessive weight gain, avoidance of long-lasting standing, and regular exercises.
Finally, we describe a rare presentation of prenatal bilateral hydrothorax evolving to hydrops fetalis. Anecdotal cases of in utero hydrothorax and nonimmune hydrops fetalis have been reported in individuals with HL-I and other primary lymphedema syndromes (Jernite et al. 1992; Lev-Sagie et al. 2003; Fang et al. 2000; Irrthum et al. 2003). VEGFR3 is known to have a major role in lymphangiogenesis, mainly of the superficial lymphatic channels (Jeltsch et al. 2003). Our report indicates that VEGFR3 may have a lesser yet nonnegligible role in the development of major lymphatic ducts, such as the thoracic duct. Reviewing our family history, including pregnancy courses and outcomes (e.g., miscarriage rate, premature births), we concluded that prenatal hydrothorax or hydrops fetalis in individuals carrying VEGFR3 mutation is a rare consequence. Interestingly, pleural effusion was noted in another family from Israel with a VEGFR3 mutation (Lev-Sagie et al. 2003; A. Ghalamkarpour et al., manuscript in preparation). In contrast with nonimmune hydrops fetalis of other causes, where outcome is usually grave, in cases with family history of Milroy disease complicated by hydrops fetalis, the outcome may be favorable providing there is early intervention.
In summary, HL-I is a disorder with broad phenotypic spectrum, even in patients bearing the same mutation. Clinical presentation ranges from asymptomatic individuals (incomplete penetrance) through “classic,” mainly cosmetic, features to late complications and rare prenatal life-threatening conditions. The interplay between genetic modifiers and environmental factors seems to determine the specific phenotype of each affected individual.
References
Brice G, Child AH, Evans A, Bell R, Mansour S, Burnand K, Safarazi M, Jeffery Sand Mortimer P (2005) Milroy disease and the VEGFR-3 mutation phenotype. J Med Genet 42:98–102
Dale RF (1985) The inheritance of primary lymphedema. J Med Genet 22:274–278
Daniel-Spiegel E, Ghalamkarpour A, Spiegel R, Weiner E, Vikkula M, Shalev E, Shalev SA (2005) Hydrops fetalis: an unusual prenatal presentation of hereditary congenital lymphedema. Prenatal Diagn 25:1015–1018
Dieppe PA, Lohmander LS (2005) Pathogenesis and management of pain in osteoarthritis. Lancet 365:965–973
Evans AL, Brice G, Sotirova V, Mortimer P, Beninson J, Burnand K, Rosbotham J, Child AH, Safarazi M (1999) Mapping of primary congenital lymphedema to the 5q35.3 region. Am J Hum Genet 64:547–555
Evans AL, Bell R, Brice G, Comeglio P, Lipede C, Jeffery S, Mortimer P, Safarazi M, Child AH (2003) Identification of eight novel VEGFR-3 mutations in families with primary congenital lymphedema. J Med Genet 40:697–703
Fang J, Dagenais SL, Erickson RP, Arlt MF, Glynn MW, Gorski JL, Seaver LH, Glover TW (2000) Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am J Hum Genet 67:1382–1388
Ferrell RE, Levinson KL, Esman JH, Kimak MA, LawrenceEC, Barmada MM, Finegold DN (1998) Hereditary lymphedema: evidence for linkage and genetic heterogeneity. Hum Mol Genet 7:2073–2078
Greenlee R, Hoyme HE, Witte MH, Crowe P, Witte CL (1993) Developmental disorders of the lymphatic system. Lymphology 26:156–168
Irrthum A, Karkkainen MJ, Devriendt K, Alitalo K, Vikkula M (2000) Congenital hereditary lymphedema caused by a mutation that inactivates VEGFR3 tyrosine kinase. Am J Hum Genet 67:295–301
Irrthum A, Devriendt K, Chitayat D, Mtthijs G, Glade C, Steijlen PM, Fryns JP, Van Steensel MA, Vikkula M (2003) Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. Am J Hum Genet 72:1470–1478
Jeltsch M, Tammela T, Alitalo K Wilting J (2003) Genesis and pathogenesis of lymphatic vessels. Cell Tissue Res 314:69–84
Jernite M, Donato L, Favre R Haddad J, Esposito M, Messer J (1992) Medical treatment of chylous effusions in newborn infants: apropos of 3 cases. Arch Fr Pediatr 49:811–814
Karkkainen MJ, Ferrell RE, Lawrence EC, Kimak MA, Levinson KL, McTigue MA, Alitalo K, Finegold DN (2000) Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet 25:153–159
Kim H, Dumont DJ (2003) Molecular mechanisms in lymphangiogenesis: model systems and implications in human disease. Clin Genet 64:282–292
Lev-Sagie A, Hamani Y, Raas-Rothschild A, Yagel S, Anteby EY (2003) Prenatal ultrasonographic diagnosis of atypical Nonne-Milroy lymphedema. Ultrasound Obstet Gynecol 21:72–74
Levinson KL, Feingold E, Ferrel RE, Glover TW, Traboulsi EI, Finegold DN (2003) Age of onset in hereditary lymphedema. J Pediatr 142:704–708
Witte MH, Erikson R, Bernas M, Andrade M, Reiser F, Conlon W, Hoyme HE, Witte CL (1998) Phenotypic and genetic heterogeneity in familial Milroy lymphedema. Lymphology 31:145–155
Acknowledgments
These studies were partially supported by Interuniversity Attraction Poles initiated by the Belgian Science Policy network 5/25, Concerted Actions Convention no. 02/07-276 of the Belgian French Community Ministry, the FNRS (Fonds National de la Recherche Scientifique) and the Integrated Project (LSHG-CT-2004-503573) of the European Commission’s Sixth Framework Programme (to MV, a Maître de recherche du FNRS).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Spiegel, R., Ghalamkarpour, A., Daniel-Spiegel, E. et al. Wide clinical spectrum in a family with hereditary lymphedema type I due to a novel missense mutation in VEGFR3 . J Hum Genet 51, 846–850 (2006). https://doi.org/10.1007/s10038-006-0031-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-006-0031-3
Keywords
This article is cited by
-
A family with Milroy disease caused by the FLT4/VEGFR3 gene variant c.2774 T > A
BMC Medical Genomics (2021)
-
Single nucleotide polymorphisms in the angiogenic and lymphangiogenic pathways are associated with lymphedema caused by Wuchereria bancrofti
Human Genomics (2017)
-
Congenital, low penetrance lymphedema of lower limbs maps to chromosome 6q16.2–q22.1 in an inbred Pakistani family
Human Genetics (2008)
