
Abstract
Three new phthalide derivatives, emefuranones A1, A2 and B (1–3); six new phthalane derivatives, emefuran A, B1, B2, C1, C2 and D (4–9); three new farnesylated phthalide derivatives, farnesylemefuranones A–C (10–12); xylarinol C (13); and emericelloxide (14), along with four known compounds (dustanin, sorbicillin, aspergillodiol and xylarinol A), were isolated from the culture extracts of Emericella sp. IFM57991. Structures of 1–14 were elucidated on the basis of spectroscopic analysis and chemical evidence. Compounds 4–7 and 13 showed moderate antibacterial activities against Bacillus subtilis.
Similar content being viewed by others
Introduction
Ascomycete fungi of the genus Emericella are well known for their ability to produce diverse bioactive compounds. Our research group previously has isolated multiple secondary metabolites (some with bioactivities) from species of this genus, with examples including emestrin (a novel antifungal macrocyclic epidithiodioxopiperazine) from E. striata1, 2 emesterones A-B3 (18,22-cyclosterols with antifungal and cytotoxic activities) and emethallicins A-F4, 5, 6 (epitetrathiodioxopiperazine derivatives that are potent inhibitors of mast cell histamine release) from E. heterothallica; variecolol, variecolactone and variecolin7 (sesterterpene inhibitors of angiotensin II receptor binding), epurpurins A-C8, 9 (dicyanide derivatives), and emindoles PA-PC10 (indoloditerpenes) from E. purpurea; falconensins A-N11, 12, 13, 14, 15 (azaphilone derivatives with anti-inflammatory activity), falconensones A-B16, 17, 18, 19 (yellow pigments with antioxidant activity) and differentiation and apoptosis inducers from E. falconensis and E. fruitculosa; and nidulalin A (a dihydroxanthone derivative, subsequently reported to have DNA topoisomerase II inhibitor activity20, 21) and nidulalin B (a benzophenone derivative, subsequently reported to have immunosuppressive activity21) from E. nidulans.22
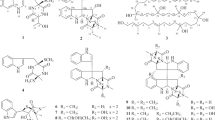
During our studies in search of bioactive compounds from fungi, we have isolated the interesting new fungal strain, IFM57991, belonging to the genus Emericella. From the culture extract of this fungus, we have isolated three new isobenzofuranone derivatives, emefuranones A1, A2 and B (1–3); six new isobenzofuran derivatives, emefuran A, B1, B2, C1, C2 and D (4–9); three new farnesylated isobenzofuranone derivatives, farnesylemefuranones A-C (10–12); xylariol C (13); and emericelloxide (14), along with four known compounds (dustanin,23 sorbicillin,24 aspergillodiol25 and xylariol A26; Figure 1).

New compounds 1–14 and their derivatives from Emericella sp. IFM57991.
In this paper, the isolation, structure elucidation and biological activities of these compounds are described.
Results and Discussion
Planar structure of emefuranones (1–3) and emefurans (4–9)
The molecular formulae of 1–9 are summarized in Supplementary Table S1. The 1H- and 13C-NMR spectra of 1–9 are shown in Table 1. A detailed analysis of the 1H-NMR spectra of 1–9 revealed that these compounds share o-couplings phenyl proton signals, two singlet methyl proton signals, an olefin proton signal and methylene proton signals couplinged above the olefin proton. 1H-1H COSY spectra showed the correlation between 5-H and 6-H in all 9 compounds (1–9), and the correlation between 1′′-H2 and 2′′-H was observed in 1 and 3 –9. The HMBC from 4′′-H3/5′′-H3 to C-2′′, C-3′′ and C-5′′/C-4′′, from 1′′-H2 to C-3a and C-5, from H-5 to C-3a, C-7, and C-1′′, and from 6-H to C-4, C-7, and C-7a were observed in the partial structures of 1–9, respectively. In addition, the 1H-1H COSY spectrum of 2 did not detect the correlation between 1′′-H2 and 2′′-H, but a HMBC correlation from 2′′-H to C-4 was shown. On the basis of the above data, we postulated that compounds 1–9 have a prenylated hydroxyphenyl moiety as part of their common partial structure (Figure 2).

Selected 1H-1H COSY and HMBC correlations of 1-9.
The molecular formulae of both 1 and 2 were established as C21H30O5, and each compound had 7 degrees of unsaturation. The 1H-NMR spectra of 1 and 2 each showed a primary methyl proton signal (1:δ 0.85 (t, J=6.2 Hz), 2: δ 0.85 (t, J=6.6 Hz) each), three methine proton signals including two oxygenated ones, five methylene proton signals and the peaks derived from the common moiety (Table 1). The 13C-NMR spectra of 1 and 2 both showed a characteristic carbonyl carbon signal (Table 1). The benzofuranone moiety in 1 and 2 was confirmed based on the correlations of HMBC spectra in 1 and 2 from 3-H to C-1, C-3a and C-7a (Figure 2). The 1H-1H COSY spectrum of 1 showed the continuous correlation between 3-H and 5′-H2, and that of 2 showed the continuous correlation between 3-H and 4′-H2 (Figure 2). The correlations of the HMBC spectrum of 1 were observed from 7′-H2 to C-5′ and C-6′ and from 8′-H3 to C-6′ and C-7′, and the correlations of the HMBC spectrum of 2 were observed from 7′-H2 to C-5′ and C-6′ and from 8′-H3 to C-6′ (Figure 2). These results suggested that the planar structure of 1 and 2 was established as 3-(2,3-dihydroxyoctyl)-7-hydoroxy-4-(3-methylbut-2-en-1-yl)-(3H)- isobenzofuran-1-one. Furthermore, 1 and 2 were hypothesized to be diastereomers based on the differences in melting point and optical rotation values when compared with each other (Supplementary Table S1).
The molecular formula of emefuranone B (3) was determined to be C15H16O5 by HR-EI-MS (Supplementary Table S1). Comparison of the 13C-NMR spectrum of 3 with that of 1 revealed that the peaks belonging to the dihydroxyoctyl moiety in 1 were absent from 3, whereas peaks corresponding to the C-2′ carbonyl group (δC 170.8) and methylene group in 3 were were absent from 1 (Table 1). These results and analysis of two-dimensional (2D)-NMR spectra established the structure of 3 as 2-[4-hydroxy-7-(3-methylbut-2-en-1-yl)-3-oxo-1,3-dihydroisobenzofuran-1-yl] acetic acid (Figure 2).
The molecular formula of emefuran A (4) was deduced as C21H32O4, that is, the molecule had one fewer oxygen atom and two more hydrogen atoms than 1, as judged by HR-CI-MS (Supplementary Table S1). The 1H- and 13C-NMR data of 4 were similar to those of 1, except for that of the C-1 (4: δ 69.3, 1: δ 168.1), and a new oxymethylene (δH 4.83 and δH 4.89) present in 4 (Table 1). These facts suggested that 4 was a 1-deoxo derivative of 1. Morever, based on the detailed analysis of 2D-NMR spectra in 4, the structure of 4 was determined to be 3-(2,3-dihydroxyoctyl)-7-hydoroxy-4-(3-methylbut-2-enyl)-(3H)-isobenzofuran (Figure 2).
Emefurans B1 (5) and B2 (6) were separated by preparative HPLC on a silica gel column, with respective retention time (tR) values of 14.5 and 20.0 min. The molecular formula of these compounds was hypothesized as C21H30O4 by HR-EI-MS and HR-CI-MS (Supplementary Table S1), and the 1H- and 13C-NMR spectra of 5 and 6 were very similar to each other (Table 1). Moreover, the 1H- and 13C-NMR spectra of 5 and 6 also were similar to those of 4, except for that of the C-2′ (δ 70.8 in 4, δ 211.0 in both 5 and 6), and the absence of 4′s methine signal at 2′-H (δ 3.52) from 5 and 6 (Table 1). Based on the above results and the detailed analysis of the 2D-NMR spectra of 5 and 6, it was hypothesized that 5 and 6 have the same planar structures, and that the two compounds were C-2′ oxygenated derivatives of 4 (Figure 2).
Emefurans C1 (7) and C2 (8) were determined to have the same molecular formulae (C23H32O5) by HR-EI-MS and HR-CI-MS, respectively (Supplementary Table S1), and the 1H- and 13C-NMR spectra of 7 and 8 were similar to each other. Furthermore, optical rotation values and CD spectra of 7 and 8 suggested that these compounds were diastereomeric with each other. Comparison of the 1H- and 13C-NMR spectra between 7 and 5, and between 8 and 6, revealed a lower field shift of a methine proton at C-3′ position (from δH 3.93 in 5 and δH 3.92 in 6 to δH 4.98 in both 7 and 8) and the new appearance of an acetyl signal (7: δH 2.07/δC 20.4 and δC 170.0; 8: δH 2.07/δC 20.4 and δC 169.9) (Table 1). Based on the above results and a detailed analysis of the 2D-NMR spectra of 7 and 8, it was suggested that 7 and 8 had the same planar structures, and that the two were acetylated derivatives of 5 and 6, respectively (Figure 2).
The molecular formula of emefuran D (9) was confirmed as C15H18O4 by HR-EI-MS (Supplementary Table S1). The 1H- and 13C-NMR spectra of 9 were similar to those of 3, except for the C-1 (δc 60.7 in 9, δc 167.8 in 3) (Table 1). Therefore, 9 was a reductant of 3 at the C-1 position. Detailed analysis of the 2D-NMR spectra confirmed that 9 was the 1-deoxo derivative of 3 (Figure 2).
Stereochemistry of emefuranones (1–3) and emefurans (4–9)
The absolute configurations at the C-2′ and C-3′ positions in 1, 2, and 4 were determined by the method of Riguera et al., employing a technique that used the α-methoxy-α-(trifluoromethyl)-phenylacetyl chloride (MTPA-Cl) reagent.27, 28 The differences in chemical shift values (Δδ = δS−δR) between diastereomeric MTPA esters (1a and 1b, 2a and 2b, 4a and 4b) were calculated to determine the absolute configuration at the C-2′ and C-3′ positions (Supplementary Figure S1). From the resulting calculated values, it was determined that the stereochemistry at the C-2′ and C-3′ positions in 1, 2 and 4 were all in the S configurations. The absolute configurations at the C-3 position in 1 and 2 were assigned by comparison of the CD spectra of 1 and 2 with that of spirolaxine.29 A cotton effect at 220 nm of 2 showed the same chirality as that of spirolaxine, and the CD spectrum of 1 displayed the opposite chirality compared with that of spirolaxine and 2 (Supplementary Figure S2). Therefore, it was determined that the absolute configuration at the C-3 position in 1 and 2 was S and R configuration, respectively.
The absolute configuration at the C-3′ position in 5 and 6 was determined to be S configuration based on the modified Mosher’s method30 (Supplementary Figure S3). To determine the absolute configurations at C-3 in 5 and 6, these compounds were used as substrates for attempted reaction via the oxidation cleavage and phenylglycine methyl ester (PGME) method.31 These compounds were treated with KIO4 to obtain the cleavage products 15 and 16, and 15 and 16 were treated with (R)- and (S)- PGME to produce the amide derivatives (15a, 15b, 16a and 16b) (Supplementary Figure S4). The absolute configuration at the C-3 positions in 15 and 16 was determined to be R and S, respectively, based on the results of the differences in chemical shift value of the PGME amide of 15 and 16 (Δvalues = δR −δS). Therefore, the absolute configuration of the C-3 position in 5 and 6 was inferred as R and S, respectively.
The CD curve of 4 was similar to that of 5, and showed opposite orientation to that of 6. Therefore, the absolute configuration at C-3 in 4 was inferred as the S configuration (Supplementary Figure S5).
To determine the stereochemistry of 7 and 8, 5-8 were acetylated with acetic anhydride. 1H-NMR and CD spectra of acetylated 7 were the same as those of the acetylated 5, and those of acetylated 8 were the same as those of acetylated 6. Therefore, we inferred that the C-3 and C-3′ positions of 7 were in the same S configuration as 5, and that those of 8 were in the same stereochemistry as 6 (C-3 and C-3′ with R and S configuration, respectively).
The CD spectra and optical rotations of emefranone B (3) and emefuran D (9) showed values of near zero. Treatment of 3 and 9 with (S)-PGME produced the PGME amide derivatives, and the results of HPLC analysis with these derivatives showed that the (S)-PGME derivatives for 3 and 9 each included two peaks (derivative of 3: tR = 23.2 and 25.1 min; derivative of 9: tR = 27.3 and 28.8 min) on their HPLC chromatograms (Supplementary Figure S6). These facts were consistent with the inference that 3 and 9 were racemic compounds.
Structure of farnesylemefuranones A–C (10–12)
Farnesylemefuranone A (10) was obtained as a colorless amorphous powder. The molecular formula was determined to be C24H32O6 on the basis of HR-ESI-MS (Supplementary Table S1). The 1H-NMR spectrum showed an aromatic proton signal (δ 6.99, s), two olefin proton signals, two oxymethylene signals, a methoxy proton signal, two singlet methyl proton signals, two doublet methyl proton signals, three sp3 methylene proton signals and a sp3 methine proton signal (Table 2). The 13C-NMR and HSQC (heteronuclear single quantum coherence) spectra assigned two carbonyl carbons (δ 214.7 and δ 171.4), 10 sp2 carbons, a methoxy carbon (δ 56.3), four methyl carbons (δ 18.3, δ 18.3, δ 16.4 and δ 16.1), six sp3 methylene carbons including two oxygenated one (δ 69.7 and δ 67.5) and a sp3 methine carbon (δ 40.8) (Table 2). The four partial structures (H-2′ to H-3′, H-5′ to H-7′, H-9′ to H-10′ and H-12′ to H-14′) were deduced from analysis of the 1H-1H COSY spectrum (Figure 3). The HMBC correlations showed the presence of an isobenzofuranone moiety and a farnesyl moiety in 12. Futhermore, we observed HMBC correlations from H2-2′ (δ 4.68) to the quaternary sp2 carbon at the C-5 position of an isobenzofuranone moiety via an ether linkage, and from the methoxy proton (δ 3.91) to the quaternary sp2 carbon at the C-6 position. These results indicated that the farnesyl moiety and methoxy group were connected to the C-5 and C-6 positions of the isobenzofuranone moiety, respectively (Figure 3). The stereochemistry of the olefin group in 10 was elucidated using selected NOESY correlations, and all double bonds were determined to be in the E configuration (Supplementary Figure S7). Therefore, the structure of 10 was established as shown in Figure 1.

Selected 1H-1H COSY and HMBC correlations of farnesylemefuranones A (10)–C (12).
The molecular formulae of farnesylemefuranone B (11) and C (12) were determined to be C23H30O6 and C23H32O7 by HR-EI-MS and HR-ESI-MS, respectively. Comparison of the 1H- and 13C-NMR spectra of 11 and 12 with those of 10 showed the presence of an isobenzofuranone moiety in 11 and 12. However, the methoxy group in 10 was absent from both 11 and 12 (Table 2). Compound 11 had five sp2 carbons, including a carbonyl carbon (δ 215.9), when compared with 10. Instead of the carbonyl carbon (δ 214.7) and methine carbon (δ 40.8) seen in 10, two oxymethines (δC 78.1 and δC 73.2) were observed in 12. Therefore, 11 and 12 were inferred to be farnesylated isobenzofuranones. Based on the above results and detailed analysis of 2D NMR spectra (Figure 2), the planar structure of 11 and 12 was determined as shown in Figure 1. The stereochemistries of the olefin groups in 11 and 12 were elucidated using selected NOESY and NOE correlations and the coupling constant, and all double bonds were determined to be in the E configuration (Supplementary Figure S7). The absolute configuration at C-11′ in 12 was elucidated to be S using the Mosher’s method (Supplementary Figure S8).
Structure of xylarinol C (13) and emericelloxide (14)
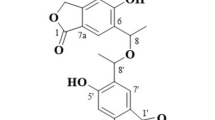
Xylarinol C (13) was obtained as pale yellow amorphous solid. The molecular formula of 13 was determined as C15H16O3 by HR-EI-MS. The 2D-NMR spectral data of 13 indicated the presence of an isoprenyl group and a 9-hydoxybenzo[c]oxepin-3(1H)-one skeleton similar to that of xylarinol A (Figure 1). Furthermore, the HMBC correlation from the H-1′ (δ 3.37) to the C-6 position (δ 131.8) on the benzene ring of 13 indicated that the isoprenyl group was connected to the C-6 position of the 9-hydoxybenzo[c]oxepin-3(1H)-one skeleton (Figure 4). Therefore, we inferred that xylarinol C (13) was a 9-isoprenyl derivative of xylarinol A.

Selected 1H-1H COSY and HMBC correlations of xylarinol C (13).
The molecular formula of emericelloxide (14) was determined as C21H26O3 by HR-ESI-MS; the 1H-NMR spectrum of 14 showed o-couplings phenyl proton signals [δ 6.67 (J = 8.4 Hz), and δ 6.89 (J = 8.4 Hz)], three olefinic proton signals (δ 5.14, δ 6.15 and δ 6.72), an oxygenated methine proton signal (δ 4.95), five methylene proton signals, a primary methyl proton signal [δ 0.87 (3H, t, J = 6.8 Hz)] and two singlet methyl proton signals. The 13C-NMR spectrum of 14 showed a carbonyl carbon signal, 10 sp2 carbon signals, two oxygenated carbon signals and eight sp3 carbon signals. These results suggested that the partial structure of 14 included a prenylated hydroxyphenyl moiety, although 14 lacked the furanone and furan moieties of 1–9. Detailed analysis of the 2D-NMR spectra indicated that the structure of 14 was 2-hexanoyl-7-hydoroxy-4-(3-methylbut-2-en-1-yl)-1,2-dihydronaphth[1,2-b]oxirene (Figure 4).
Antifungal and antibacterial activities and cytotoxicity were tested for 1–9, 13, 16, 17, dustanin and sorbicillin. Compounds 4–7 and 13, 16 and 17 showed weak inhibition of B.subtilis (respective inhibition zones: 8.5, 10, 9, 9, 13, 9 and 9 mm in 250 μg per disk). Cytotoxicity and antifungal activity were not observed at concentrations of 50 μM and 250 μg per disk, respectively.
Isobenzofuranone derivatives, emefuranones A1, A2, B (1–3), isobenzofuran derivatives emefurans A, B1, B2, C1, C2, D (4–9), and farnesylemefuranones A-C (10–12), xylariol C (13) and emericelloxide (14) were isolated, along with known compounds dustanin, sorbicillin, aspergillodiol and xylarinol A from the culture extract of Emericella sp. IFM57991. Compounds 4–7, 13, 16 and 17, showed moderate antibacterial activity against B. subtilis and no activities against Escherichia coli.
Materials and Methods
General procedure
Melting points were measured on a Yanagimoto micro-melting point apparatus (Yanaco New Science Inc., Kyoto, Japan). Optical rotations were measured on a JASCO DIP-1000 (JASCO Co., Ltd., Tokyo, Japan). CD spectra were obtained with a JASCO J-820 spectrophotometer (JASCO Co., Ltd.). UV spectra were recorded on an Amersham Biosciences, Ultrospec 2100 spectropolarimeter (GE Healthcare Japan Corp., Tokyo, Japan). Measurements of IR spectra were performed with a JASCO FT/IR-4100 spectrometer on a KBr cell (JASCO Co., Ltd.). NMR spectra (both 1 and 2D) were obtained on a Bruker AVANCE-400 (400.13 MHz for 1H and 100.61 MHz for 13C) spectrometer (Bruker Bio Spin K. K., Kanagawa, Japan). Multiplicity of signals is abbreviated as follows: s, singlet; d, doublet; dd, doublet of doublet; dt, doublet of triplets; t, triplet; q, quartet; m, multiplet; br, broad. EI and CI-MS spectra were obtained from a JEOL JMS-600W spectrometer (JEOL Ltd., Tokyo, Japan). ESI-MS spectra were obtained from a JEOL T100LP spectrometer (JEOL Ltd.).
Fungal strain
The fungal strain IFM57991 was obtained from the Medical Mycology Research Center, Chiba University, Japan. Colonies, which were pale brown in color, grew moderately on potato dextrose agar and produced curved hull cells. However, conidiogenesis and sexual spore formation were not observed, precluding morphological identification of the fungus at the species-level. DNA sequence analysis of the β-tubulin-encoding gene identified the strain as Emericella sp.
Culture, extraction and isolation
Culture medium consisted of moist rice, autoclaved at 120 °C for 20 min before inoculation. Emericella sp. IFM57991 was inoculated into each of 25 Roux flasks (containing 150 g of moist rice per flask) and cultured at 25 °C for 21 days. The fermented rice was extracted with MeOH and the organic layer was evaporated. The resultant extract was suspended in H2O and extracted with EtOAc, and then the organic layer was evaporated in vacuo. The EtOAc extract (11.3 g) was suspended in n-hexane and extracted with MeCN and then the MeCN layer was evaporated in vacuo. The MeCN extract (4.5 g) was chromatographed using a Sephadex LH-20 column (solvent system: hexane-CHCl3 (1:4; 200 ml), CHCl3-acetone (3:2; 200 ml), (1:4; 200 ml), acetone (200 ml) and MeOH (500 ml)) to yield 11fractions.
The third fraction was suspended in MeOH and filtered. The residual solid was confirmed to contain dustanin (200 mg). The filtrate was purified by middle-pressure liquid chromatography (MPLC) on a silica gel column (φ25x250, YAMAZEN, Osaka, Japan) using CHCl3-acetone (15:1) followed by further purification by high-performance liquid chromatography (HPLC) on a silica gel column (φ10x250, GL science, Tokyo, Japan); elution with hexane-EtOAc (5:1) yielded farnesylemefuranone A (10: 3 mg).
The fourth fraction was suspended in MeOH and filtered. The residue was confirmed to contain emefuranone A1 (1: 40 mg). The filtrate was chromatographed by MPLC on a silica gel column using CHCl3-acetone (10:1) followed by further purification by HPLC on a silica gel column. Elution with benzene-EtOAc (12:1) and then CHCl3-acetone (25:1) yielded emefuran C2 (8: 3 mg); subsequent elution with CHCl3-acetone (20:1) followed by CHCl3-EtOAc (4:1) yielded emefuranone A2 (2; 3 mg); and subsequent elution with hexane-EtOAc (4:1) yielded emericelloxide (14; 4 mg).
The fifth fraction was suspended by MeOH and filtered. The residue was confirmed to contain emefuranone A1 (1: 30 mg). The filtrate was purified by MPLC on a silica gel column using benzene-acetone (5:1) followed by further purification by HPLC on a silica gel column; elution with CHCl3-acetone (15:1) yielded emefuran C1 (7: 10 mg).
The sixth fraction was chromatographed by MPLC on a silica gel column using CHCl3-MeOH (10:1) followed by further purification by HPLC on a silica gel column. Elution with benzene-acetone (15:1) yielded sorbicillin (10 mg), xylarinol C (13; 3 mg), emefuran B1 (5; 25 mg) and emefuran B2 (6; 30 mg); subsequent elution with CHCl3-acetone (5:1) and CHCl3-EtOAc (5:1) yielded aspergillodiol (3 mg) and emefuran A (4; 4 mg); and subsequent elution with CHCl3-EtOAc (5:1) yielded farnesylemefuranone B (11; 2 mg).
The seventh fraction was purified by MPLC on a silica gel column using hexane-acetone (2:1) and benzene-acetone (4:1) followed by further purification by HPLC on a silica gel column; elution with CHCl3-MeOH (10:1) yielded emefuranone B (3: 3 mg). The residue was chromatographed by HPLC on a silica gel column; elution with CHCl3:acetone (5:1) and CHCl3:EtOAc (5:1) yielded farnesylemefuranone C (12: 2 mg).
The eighth fraction was chromatographed and purified by HPLC on a silica gel column using n-hexane-acetone (3:2) and benzene-acetone (4:1) to give emefuran D (9: 4 mg). The residue was purified by MPLC on a silica gel column using CHCl3-MeOH (15:1) followed by further purification by HPLC on a silica gel column; elution with CHCl3-EtOAc (5:1) and CHCl3-acetone (15:1) yielded xylarinol A (2 mg).
Physicochemical properties of 1–14
The physicochemical properties of new compounds (1–14) are summarized in Supplementary Table S1, and the 1H- and 13C-NMR spectral spectral data of 1-12 are shown in Tables 1 and 2.
Xylariol C (13)
1H-NMR (DMSO-d6) δ 1.71 (3H, s, H3-4′), 1.73 (3H, s, H3-5′), 3.37 (2H, d, J = 7.1 Hz, H-1′), 5.15 (2H, t, J = 7.1 Hz, H-2′), 5.22 (2H, brs, H-1), 6.34 (1H, d, J = 12,3 Hz, H-4), 6.89 (1H, d, J = 8.4 Hz, H-8), 7.12 (1H, d, J = 8.4 Hz, H-7) and 7.46 (1H, d, J = 12.3 Hz, H-5). 13C NMR (MeOD) δ 16.6 (C-5′), 24.4 (C-4′), 31.1 (C-1′), 61.6 (C-1), 116.7 (C-8), 121.1 (C-4), 122.2 (C-9a), 122.6 (C-2′), 130.8 (C-7), 131.8 (C-6), 132.2 (C-3′), 134.7 (C-5a), 138.6 (C-5), 150.8 (C-9) and 169.8 (C-3).
Emericelloxide (14)
1H-NMR (DMSO-d6) δ 0.87 (3H, t, J = 6.8 Hz, 6′-H), 1.26 (2H, m, 4′-H2), 1.28 (2H, m, 5′-H2), 1.45 (2H, m, 3′-H2), 1.67 (3H, s, 4′′-H3), 1.69 (3H, s, 5′′-H3), 2.57 (1H, dt, J = 17.8, 7.6 Hz, 2′-H2), 2.84 (1H, dt, J = 17.8, 7.6 Hz, 2′-H2), 3.26 (2H, t, J = 7.4 Hz, 1′′-H2), 4.95 (1H, s, 1-H), 5.14 (1H, t, J = 7.4 Hz, 2′′-H), 5.50 (1H, s, 8-OH), 6.15 (1H, d, J = 10.1 Hz, 3-H), 6.67 (1H, d, J = 8.4 Hz, 7-H), 6.72 (1H, d, J = 10.1 Hz, 4-H) and 6.89 (1H, d, J = 8.4 Hz, 6-H). 13C-NMR (DMSO-d6) δ 13.9 (C-6′), 17.7 (C-5′′), 22.0 (C-5′), 22.8 (C-3′), 25.5 (C-4′′), 30.3 (C-1′′), 30.9 (C-4′), 35.9 (C-2′), 66.4 (C-1), 75.6 (C-2), 115.4 (C-7), 120.7 (C-8a), 124.0 (C-2′′), 125.3 (C-4), 127.7 (C-3), 127.8 (C-5), 129.4 (C-6), 130.0 (C-4a), 130.6 (C-3′′), 153.8 (C-8) and 210.1 (C-1′). The chemical shift of C-3 may be exchanged with C-5.
Esterification of emefuranone A1 (1), A2 (2), emefuran A (4), B1 (5) and B2 (6) with MTPA-Cl
Well-dried samples (1: 2 mg, 2: 1 mg, 4: 1 mg, 5: 2 mg, 6: 2 mg) were dissolved in pyridine-d5 (1: 200 μl, others: 300 μl) and added to (R)-(-)-MTPA-Cl (150 μl) or (S)-(+)-MTPA-Cl (150 μl) in each NMR tube (φ 2.5 mm). Then, the NMR tubes were held for 15 h at 50 °C. The assignment was made on the basis of 1H-NMR spectrum.
Oxidative cleavage of emefuran B1 (5) or emefuran B2 (6)
Emefuran B1 (5: 6 mg) or emefuran B2 (6: 5 mg) were stirred with KIO4 (15 mg) in H2O (0.5 ml) and dioxane (0.5 ml) for 12 h at 50 °C. Then, H2O (10 ml) was added, the suspension was extracted with diethyl ether, and the ether layer was evaporated. After concentration in vacuo, the organic layer was chromatographed and purified by HPLC on silica gel column using hexane-acetone (3:2) to give 15 (1 mg) or 16 (0.5 mg).
Compound 15
Colorless amorphous powder. ESI-MS m/z: 285 (M+Na)+. UV λmax (MeOH) nm (log ɛ): 213 (4.6), 221 (4.2) and 280 (3.4). CD (MeOH) Δɛ (nm): 6.1 (209), -1.5 (231), -0.6 (276). H-NMR (CDCl3) δ 1.70 (3H, s, 5′′-H), 1.73 (3H, s, 4′′-H), 2.61 (1H, dd, J = 15.8, 9.6 Hz, 1′-H), 2.89 (1H, dd, J = 15.8, 2.2 Hz, 1′-H), 3.22 (2H, t, J = 7.1 Hz, 1′′-H), 5.11 (1H, d, J = 12.4 Hz, 1-H), 5.18 (1H, m, 2′′-H), 5.20 (2H, m, 1-H), 5.73 (1H, brd, J = 9.6 Hz, 3-H), 6.64 (1H, d, J = 8.2 Hz, 6-H) and 6.96 (1H, d, J = 8.2 Hz, 5-H).
Compound 16
Colorless amorphous powder. ESI-MS m/z: 285 (M+Na)+. UV λmax (MeOH) nm (log ɛ): 212 (4.3), 223 (4.2) and 277 (3.5). CD (MeOH) Δɛ (nm): -4.4 (212), 1.4 (230), 0.6 (276). H-NMR (CDCl3) δ 1.70 (3H, s, 5′′-H), 1.73 (3H, s, 4′′-H), 2.61 (1H, dd, J = 15.8, 9.6 Hz, 1′-H), 2.89 (1H, dd, J = 15.8, 2.2 Hz, 1′-H), 3.22 (2H, t, J = 7.1 Hz, 1′′-H), 5.11 (1H, d, J = 12.4 Hz, 1-H), 5.18 (1H, m, 2′′-H), 5.20 (2H, m, 1-H), 5.73 (1H, brd, J = 9.6 Hz, 3-H), 6.64 (1H, d, J = 8.2 Hz, 6-H) and 6.96 (1H, d, J = 8.2 Hz, 5-H).
Acetylation of emefuran B1 (5), emefuran B2 (6), emefuran C1 (7) and emefuran C2 (8)
Emefuran B1 (5: 2 mg), emefuran B2 (6: 2 mg), emefuran C1 (7: 2 mg) or emefuran C2 (8: 2 mg) was dissolved in pyridine (300 μl) in each Erlenmeyer flask. Acetic anhydride (200 μl) then was added to each flask and reactions were held at room temperature overnight. After the reaction, the reactants were added to H2O and extracted with EtOAc. The organic layer was evaporated, and then purified by HPLC on a silica gel column using CHCl3-EtOAc (40:1) to obtain 17 (each 1.2 and 0.7 mg) from 5 and 7, or to obtain 18 (1.7 and 0.7 mg) from 6 and 8.
Compound 17
Colorless amorphous powder. ESI-TOF-MS m/z: 453.2292 [(M+Na)+ 453.2253 for C25H34O6Na]. UV λmax (MeOH) nm (log ɛ): 206 (4.6), 219 (4.3). CD (MeOH) Δɛ (nm): 2.1 (206), -1.1 (229), 0.7 (277). H-NMR (DMSO-d6) δ 0.86 (3H, t, J = 6.7 Hz, 8′-H), 1.17-1.38 (6H, m, 5′-, 6′-, 7′-H), 1.58-1.77 (2H, m, 4′-H), 1.65 (3H, s, 5′′-H), 1.70 (3H, s, 4′′-H), 2.08 (3H, s, 3’-OAc), 2.25 (3H, s, 7-OAc), 2.76 (1H, dd, J = 17.2, 1.6 Hz, 1′-H), 2.98 (1H, dd, J = 17.2, 9.6 Hz, 1′-H), 3.25 (2H, d, J = 7.0 Hz, 1′′-H), 4.80 (1H, d, J = 12.8 Hz, 1-H), 4.90 (1H, dd, J = 12.8, 2.3 Hz, 1-H), 4.99 (1H, dd, J = 8.5, 3.8 Hz, 3′-H), 5.17 (1H, t, J = 7.1 Hz, 2′′-H), 5.69 (1H, dt, J = 9.6,1.6 Hz, 3-H), 6.99 (1H, d, J = 8.2 Hz, 6-H), 7.10 (1H, d, J = 8.2 Hz, 5-H).
Compound 18
Colorless amorphous powder. ESI-TOF-MS m/z: 453.2291 [(M+Na)+, 453.2253 for C25H34O6Na). UV λmax (MeOH) nm (log ɛ): 206 (4.4), 219 (4.1). CD (MeOH) Δɛ (nm): -2.5 (202), 1.7 (229), 1.4 (289). H-NMR (DMSO-d6) δ 0.85 (3H, t, J = 6.7 Hz, 8′-H), 1.21–1.34 (6H, m, 5′-, 6′-, 7′-H), 1.62–1.78 (2H, m, 4′-H), 1.64 (3H, s, 5′′-H), 1.70 (3H, s, 4′′-H), 2.07 (3H, s, 3′-OAc), 2.25 (3H, s, 7-OAc), 2.70 (1H, dd, J = 17.1, 2.0 Hz, 1′-H), 2.96 (1H, dd, J = 17.1, 9.2 Hz, 1′-H), 3.23 (2H, d, J = 6.7 Hz, 1′′-H), 4.80 (1H, d, J = 12.9 Hz, 1-H), 4.91 (1H, dd, J = 12.9, 2.4 Hz, 1-H), 4.98 (1H, dd, J = 8.2, 4.5 Hz, 3′-H), 5.17 (1H, brt, J = 6.7 Hz, 2′′-H), 5.67 (1H, brd, J = 9.2 Hz, 3-H), 6.99 (1H, d, J = 8.1 Hz, 6-H) and 7.10 (1H, d, J = 8.1 Hz, 5-H).
Amidation of emefuranone B (3) with PGME-Cl
Emefuranone B (3: 1 mg) was treated with (S)-(+)-PGME-Cl (20 mg), PyBOP (22 mg), HOBT (13.0 mg) and NMM (100 μl) in DMF (1 ml) at room temperature for 20 h. EtOAc and 6% HCl aqueous solution (25 ml) were added; the EtOAc layer was washed with saturated NaHCO3 aqueous solution (25 ml); and the resulting EtOAc layer was added to 4% NaCl aqueous solution (25 ml) and partitioned. The organic layer was desiccated with anhydrous sodium sulfate. After filtration and evaporation, the extract was chromatographed by HPLC on an ODS column (φ10x250, GL science, Japan) using 60% MeOH.
Amidation of emefuran D (9) with PGME-Cl
Emefuran D (9: 1 mg) was treated with (S)-(+)-PGME-Cl (20 mg), PyBOP (22 mg), HOBT (13.0 mg) and NMM (100 μl) in DMF (1 ml) at room temperature for 20 h. EtOAc and 6% HCl aqueous solution (25 ml) were added, and the EtOAc layer was washed with saturated NaHCO3 aqueous solution (25 ml); the resulting EtOAc layer was added to 4% NaCl aqueous solution (25 ml), and partitioned. The organic layer was desiccated with anhydrous sodium sulfate. After filtration and evaporation, the extract was chromatographed by HPLC on an octadecylsilyl (ODS) column using 60% MeOH.
Cytotoxicity assay
Cytotoxicity assays were performed by a modification of the previously described method32). Cells were distributed into 96-well microplates at 4000 cells per well and allowed to attach for 4–6 h. Then, cells were grown to 80% confluency in respective media as follows: A549 human lung cancer cells and Hela human cervical cancer cells were incubated in Dulbecco’s modified Eagle’s medium (Invitrogen Co., Ltd., Carlsbad, CA, USA); LNCap human prostate adenocarcinoma cells were incubated in RPMI-1640 medium (Wako Pure Chemical Industries, Ltd., Osaka, Japan) supplemented with 10% fetal bovine serum, penicillin G (100 U ml−1), streptomycin (100 mg ml−1) and amphotericin B (0.25 mg ml−1). Then, media were supplemented with the 419 indicated concentrations of the test compounds for 48–72 h. Cell proliferation was 420 measured using the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) to count living 421 cells by combination staining with WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium) and 1-methoxy PMS (1-methoxy-5-methylphenazinium methysulfate). Briefly, the medium was removed and replaced with 10 ml per well of Cell Counting Kit-8 solution (Dojindo Molecular Technologies, Inc., Kumamoto, Japan), and the plates were incubated for 4 h. Cell number was determined by scanning with a Bio-Rad Model Q4 550 microplate reader at 450 nm (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Antifungal assay
Antifungal assays were performed by the paper disc method using pathogenic filamentous fungi, Aspergillus fumigatus IFM 41243 and A. niger IFM 41398, and pathogenic yeasts, Candida albicans IFM 40009 and Cryptococcus neoformans ATCC 90112. Test substances, dissolved in CHCl3, were added to paper discs (8 mm), which then were dried and placed on the assay plates. The plates were incubated at 25 °C for 48–72 h and the diameters of the inhibitory zones were measured.
Antibacterial assay
Antibacterial assays were performed by the paper disc method against Bacillus subtilis ATCC6633 and Escherichia coli JCM1640. Test substances, dissolved in CHCl3, were added to paper discs (8 mm), which then were dried and placed on the assay plates. The plates were incubated at 34 °C for 24 h and the diameters of the inhibitory zones were measured.
References
Seya, H., Nakajima, S., Kawai, K. & Udagawa, S. Structure and absolute configuration of emestrin, a new macrocyclic epidithiodioxopiperazine from Emericella striata. J. Chem. Soc., Chem. Commun. 1985, 657–658 (1985).
Seya, H., Nozawa, K., Nakajima, S., Kawai, K. & Udagawa, S. Studies on fungal products. Part 8. Isolation and structure of emestrin, a novel antifungal macrocyclic epidithiodioxopiperazine from Emericella striata. X-Ray molecular structure of emestrin. J. Chem. Soc., Perkin Trans 1, 109–116 (1986).
Hosoe, T., Sameshima, T., Dobashi, K. & Kawai, K. Structures of two new 18,22-cyclosterols, emesterones A and B, from Emericella heterothallica. Chem. Pharm. Bull. 46, 850–852 (1998).
Kawahara, N., Nakajima, S., Yamazaki, M. & Kawai, K. Structure of a novel epidithiodioxopiperazine, emethallicin A, a potent inhibitor of histamine release from Emericella heterothallica. Chem. Pharm. Bull. 37, 2592–2595 (1989).
Kawahara, N., Nozawa, K., Yamazaki, M., Nakajima, S. & Kawai, K. Structures of novel epipolythiodioxopiperazines, emethallicins B, C, and D, potent inhibitors of histamine release, from Emericella heterothallica. Chem. Pharm. Bull. 38, 73–78 (1990).
Kawahara, N., Nozawa, K., Yamazaki, M., Nakajima, S. & Kawai, K. Novel epidithiodioxopiperazines, emethallicins E and F, from Emericella heterothallica. Heterocycles 30, 507–515 (1990).
Takahashi, H., Hosoe, T., Nozawa, K. & Kawai, K. Two new sesterterpenes from the ascomycetous fungus Emericella purpurea. J. Nat. Prod. 62, 1712–1713 (1999).
Takahashi, H., Nozawa, K. & Kawai, K. Isolation and structures of dicyanide derivatives, epurpurins A to C, from Emericella purpurea. Chem. Pharm. Bull. 44, 2227–2230 (1996).
Kawai, K., Sugihara, Y., Kitagawa, A. & Kawai, K. The respiration-impairing effects of epurpurin A, B and C on isolated mitochondria. J.Toxicol. Sci. 23 (suppl 2), 395 (1998).
Hosoe, T., Itabashi, T., Kobayashi, N., Udagawa, S. & Kawai, K. Three new types of indoloditerpenes, emindole PA-PC, from Emericella purpurea. Revision of the structure of emindole PA. Chem. Pharm. Bull. 54, 185–187 (2006).
Ogasawara, N. & Kawai, K. Hydrogenated azaphilones from Emericella falconensis and E. fruticulosa. Phytochemistry 47, 1131–1135 (1998).
Itabashi, T. et al. Falconensins A, B, C, and D, new compounds related to azaphilone, from Emericella falconensis. Chem. Pharm. Bull. 40, 3142–3144 (1992).
Itabashi, T., Nozawa, K., Nakajima, S. & Kawai, K. A new azaphilone, falconensin H, from Emericella falconensis. Chem. Pharm. Bull. 41, 2040–2041 (1993).
Itabashi, T., Ogasawara, N., Nozawa, K. & Kawai, K. Isolation and structures of new azaphilone derivatives, falconensins E-G, from Emericella falconensis and absolute configurations of falconensins A-G. Chem. Pharm. Bull. 44, 2213–2217 (1996).
Yasukawa, K., Itabashi, T., Kawai, K. & Takido, M. Inhibitory effects of falconensins on 12-O-tetradecanoylphorbol-13-acetate-induced inflammatory ear edema in mice. J. Nat. Med 62, 384–386 (2008).
Ogasawara, N., Mizuno, R. & Kawai, K. Structures of a new type of yellow pigments, falconensones A and B, from Emericella falconensis. J. Chem. Soc., Perkin Trans 1, 2527–2530 (1997).
Takahashi, N., Iwahori, A., Kawai, K. & Fukui, T. Induction of differentiation in human promyelocytic leukemia cell line HL60 by a new type of polyenes, falconensone A and its derivatives. Arch. Biochem. Biophys. 360, 113–120 (1998).
Takahashi, N., Tamagawa, K., Kawai, K. & Fukui, T. Antioxidant properties of a new type of polyene, falconensone A and its derivatives. Biol. Pharm. Bull. 23, 989–994 (2000).
Takahashi, N., Kubo, Y., Iwahori, A., Kawai, K. & Fukui, T. Induction of apoptosis in the human promyelocytic leukemia cell line HL60 by falconensone A and its derivatives, new polyenes. Biol. Pharm. Bull. 23, 748–754 (2000).
Sato, S. et al. Inhibition of DNA topoisomerases by nidulalin A derivatives. Bio. Pharm. Bull 23, 511–512 (2000).
Fujimoto, H., Asai, T., Kim, Y.-P. & Ishibashi, M. Nine constituents including six xanthone-related compounds isolated from two ascomycetes, Gelasinospora santi-florii and Emericella quadrilineata, found in a screening study focused on immunomodulatory activity. Chem. Pharm. Bull. 54, 550–553 (2006).
Kawahara, N., Sekita, S., Satake, M., Udagawa, S. & Kawai, K. Structures of a new dihydroxanthone derivative, nidulin A, and a new benzophenone derivative, nidulalin B, from Emericella nidulans. Chem. Pharm. Bull. 42, 1720–1723 (1994).
Tsuda, Y. & Isobe, K. Dustanin: A new pentacyclic triterpenoid as a fungal metabolite. Tetrahedron Lett. 37, 3337–3343 (1965).
Du, L. et al. Cytotoxic sorbicilllinoids and bisorbicillinoids from a marine-derived fungus Trichoderma sp. Chem. Pharm. Bull. 57, 220–223 (2009).
Lin, W. H., Li, J., Fu, H. Z. & Proksch, P. Four novel hydropyranoindeno- derivatives from marine fungus Aspergillus versicolor. Chin. Chem. Lett. 12, 435–438 (2001).
Lee, I. et al. Xylarinols A and B, two new 2-benzoxepin derivatives from the fruiting bodies of Xylaria polymorpha. J. Antibiot. 62, 163–165 (2009).
Seco, J. M., Quinoa, E. & Riguera, R. The assignment of absolute configuration by NMR. Chem. Rev. 104, 17–117 (2004).
Freire, F., Seco, J. M., Quinoa, E. & Riguera, R. Determining the absolute stereochemistry of secondary/Secondary diols by 1H NMR: Basis and applications. J. Org. Chem. 70, 3778–3790 (2005).
Bava, A., Clericuzio, M., Giannini, G., Malpezzi, L., Meille, S. V. & Nasini, G. Absolute configuration of the fungal metabolite spirolaxine. Eur. J. Org. Chem 11, 2292–2296 (2005).
Otani, I., Kusumi, T., Kashman, Y. & Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 113, 4092–4096 (1991).
Yabuuchi, T. & Kusumi, T. Phenylglycine methyl ester, a useful tool for absolute configuration determination of various chiral carboxylic acids. J. Org. Chem. 65, 397–404 (2000).
Wakana, D. et al. The cytotoxic and antifungal activities of two new sesquiterpenes, malfilanol A and B, derived from Malbranchea filamentosa. J. Antibiot. 62, 217–219 (2009).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Saito, T., Itabashi, T., Wakana, D. et al. Isolation and structure elucidation of new phthalide and phthalane derivatives, isolated as antimicrobial agents from Emericella sp. IFM57991. J Antibiot 69, 89–96 (2016). https://doi.org/10.1038/ja.2015.85
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2015.85
This article is cited by
-
Production and Optimization of Physicochemical Parameters of Cellulase Using Untreated Orange Waste by Newly Isolated Emericella variecolor NS3
Applied Biochemistry and Biotechnology (2017)
