
Abstract
Rocheicoside A (3), a nucleoside analog possessing a novel 5-(hydroxymethyl)-5-methylimidazolidin-4-one substructure, was isolated from marine-derived actinomycete Streptomyces rochei 06CM016, together with a new (4) and three known compounds. Structures of the new metabolites were elucidated by one-dimensional (1H and 13C) and 2D NMR (COSY, HMQC and HMBC) and HR-TOF-MS analyses. All the metabolites exhibited significant antimicrobial activity. A plausible mechanism was proposed for compound 3’s formation from amicetin.
Similar content being viewed by others
Introduction
Antibiotics are natural compounds produced by microorganisms as secondary metabolites to kill or inhibit other microorganisms. As their first discovery in the middle of the twentieth century, they had an important role in the treatment of infectious diseases. Up to now, pharmaceutical industry has primarily targeted drugs from soil organisms, and of the antibiotics in clinical use, most are of bacterial or fungal origin. Among the bacteria, Actinobacteria are particularly noteworthy, as they produce antibiotics such as streptomycin, neomycin, chloramphenicol, chlortetracycline and erithyromycin.1, 2 Members of Actinomycetes, especially Streptomyces, are fruitful organisms, producing ~80% of the antibiotics.3, 4
The acquired resistance of pathogen microorganisms due to their high mutation rates, toxic effects of some known antibiotics on human health and the existence of naturally resistant bacteria require the discovery of novel antibiotics.5, 6 As antibiotic discovery efforts result in identified secondary metabolite frameworks with known mode of actions, recently, the researchers have started to focus on microorganisms living in extreme conditions, namely ocean and ocean bottoms, to discover novel chemical entities.2, 7, 8 It is thought that these novel compounds in general are produced as response to stress conditions.9 This research line led to the discovery of many new microbial species producing bioactive compounds.10, 11 Recent studies have revealed that unusual marine Actinomycetes are good candidates for the discovery of new natural products.12
As part of our ongoing studies,13 the aim of this study was to discover new antimicrobial molecules from marine Actinomycetes living in unexplored areas of Mediterranean Sea.
Results and Discussion
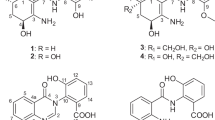
The isolation process performed using the sediment sample resulted in acquiring a bioactive mesophilic actinomycete 06CM016. The isolate showed antibacterial activity against Escherichia coli (17 mm) and MRSA (22 mm), and antifungal activity against Candida albicans (37 mm). Sequencing using primers 27F and 1492R revealed that the isolate was a member of Streptomyces genus. When the molecular identification evaluated with the phenotypical results (data not shown), the isolate was identified as Streptomyces rochei. After large-scale fermentation, 1.92 g of EtOAc extract was acquired. Bioassay-guided fractionation using the extract led to the isolation of five compounds (1–5), of which 1 (plicacetin), 2 (norplicacetin) and 5 were identified as cytosine-type nucleoside derivatives, by comparison of their spectral data with those previously reported (Figure 1).14, 15, 16, 17

Structures of 1–5.
The high resolution time of flight mass spectrum (HR-TOF-MS) of 3  (c 0.0035, MeOH)] gave molecular ions of m/z 631.3123 [(+) mode] and 629.2387 [(−) mode], attributed to [M+H]+ and [M−H]−, respectively. Together with the 13C NMR data, accurate mass data supported a molecular formula of C30H42N6O9. Initial inspection of the one-dimensional NMR spectra (1H, 13C and DEPT) also suggested a cytosine derivative, similar with compounds 1 and 2.
(c 0.0035, MeOH)] gave molecular ions of m/z 631.3123 [(+) mode] and 629.2387 [(−) mode], attributed to [M+H]+ and [M−H]−, respectively. Together with the 13C NMR data, accurate mass data supported a molecular formula of C30H42N6O9. Initial inspection of the one-dimensional NMR spectra (1H, 13C and DEPT) also suggested a cytosine derivative, similar with compounds 1 and 2.
1H NMR spectrum of 3 exhibited a tertiary and two secondary up-field methyl groups (δ 1.10, 1.16 and 1.27, respectively), a tertiary lower field methyl group (δ2.38, 6H) attributable to a N(CH3)2 unit, a para-disubstituted aromatic ring (δ 7.79, 2H, d, J=8.8 Hz; δ 8.07, 2H, d, J=8.8 Hz) and two vinyl protons (δ 7.34, 1H, d, J=7.6 Hz; δ 8.16, 1H, d, J=7.6 Hz; Table 1). The chemical shifts of the latter protons are typical of the CH(5) and CH(6) resonances of a cytosine unit.18, 19, 20 On the basis of the HMQC connectivity, this assumption was substantiated, which indicated characteristic C-5 and C-6 signals at 97.1 and 145.7 p.p.m., respectively.
In addition, 1H NMR spectrum of 3 displayed typical signals for two sugar units, namely amicetose and amosamine as in 1 and 2.16, 17, 18, 19, 20 The two major spin systems of the disaccharidic part were deduced from the COSY spectrum starting from the well resolved anomeric protons (Figure 2). The H-1′ resonance of amicetose (δ 5.73) showed cross peaks with two methylene protons (H-2′: δ 1.73 and 2.07), which, in turn, coupled with H2-3′ (δ 1.50 and 2.29). H2-3′ exhibited correlation with H-4′ (δ 3.36), whereas the latter proton showed cross peaks with H-5′ (δ 3.65), which coupled with an up-field methyl signal at δ 1.16 (H3-6′). In a similar manner, the spin system of amosamine moiety could be traced from the low-field H-1″ (δ 4.81) to H3-6″ (1.27; Table 1). The use of HMQC spectrum of 3 allowed the assignment of the carbon signals ascribable to the saccharides, which also verified the presence of inner amicetose and terminal amosamine moieties. The absolute configurations of these deoxy sugars were suggested to be D based on the same biosynthetic origin as in 1, 2 and 5. This assumption was substantiated by carrying out acidic hydrolysis on compound 3.

Spin systems and key long-range correlations of 3.
The interfragment relationship deduced from the HMBC spectrum helped us to locate the O- and N-glycosidic linkages, shown in Figure 2. Thus, the anomeric proton of the amosamine unit (δ 4.81) displayed a long-range correlation to C-4′ of amicetose (δ 73.6), whereas the anomeric carbon of the latter sugar showed a 3J C–H heteronuclear correlation with H-6 of cytosine moiety (δ 8.16). Moreover, the methyl signal observed at δ 2.38 (6H) was readily assigned to C-4″-[N(CH3)2] on the basis of a long-range connectivity to C-4″ (δ 70.5).
The combined use of COSY, HMQC and HMBC spectra also permitted the complete assignment of the aromatic ring (Table 1,Figure 2) as p-substituted benzoyl extending from cytosine moiety. The proton chemical shift of H-11(13) was notably deshielded compared with those in compound 1 (δ 7.79 and 6.57, respectively), implying an electron-withdrawing group at p-position instead of an electron donating amino group of p-amino benzoic acid, possibly an amide functionality.
After exclusion of the above-mentioned parts, the remaining side chain of the molecule had the formula C5H9N2O2. On the basis of the comprehensive examination of one-dimensional and 2D NMR data of 3, and the comparison of these data with those of related metabolites, well-known cytosine-type antibiotics, namely amicetin, bamicetin and oxamicetin,16, 19, 20, 21, 22 it was inferred that a framework derived from α-methylserine was connected to the benzoyl unit via an amide bond. Four of the left over carbons at δ 176.9, 66.2, 65.6, 19.3 (C-16 (s), C-19 (t), C-17 (s), and C-20 (q), respectively) in the 13C NMR spectrum, a singlet methyl at δ 1.10 (H3-20) and two methylene protons at δ 3.32 and 3.62 (each d, J=5.6 Hz, H2-19) in the 1H NMR spectrum were evident for the integration of the aforementioned amino-acid moiety. Furthermore, in the HMBC spectrum, long-range correlations between C-16/C-17 and H3-20/H2-19 substantiated our assumption. The remaining NMR signals indicated an isolated methylene group (H2-21: δ 4.73, 4.85, each d, J=7.6 Hz; C-21: δ 63.1, t), secured by the COSY and HMQC spectra. One of the two unsaturation numbers calculated for the side chain was readily accounted for the amide carbonyl at δ 176.9, suggestive of a cyclization. Thus, incorporation of the remaining carbon should afford a ring system. In this case, there are two possibilities, either –N–CH2–N– or –N–CH2–O– bridges. In case of the latter (oxazolidine ring), C-21 was supposed to be resonated around 80–85 p.p.m. in the 13C NMR spectrum.23, 24, 25 However, the observed carbon data (δ 63.1) was entirely compatible with –N–CH2–N– connection,26 referring a 5-(hydroxymethyl)-5-methylimidazolidin-4-one skeleton. The stereochemistry of C-17 was assigned by a combination of 2D NOESY data and molecular modeling studies performed on 3. In regard to the absolute configuration of C-17, the minimum energy conformations of R and S stereoisomers were calculated with a three-dimensional computer-generated model. In the case of R-configuration at C-17 (d-serine derivative), CH3-20 is required to show NOE interactions with CH2-21 due to their close proximity (2.64 Å) above the rigid imidazolidin-4-one ring system, whereas the S-configuration (l-serine) is predicted to provide a correlation between CH2-19 and CH2-21 (2.58 Å) (calculated CH3-20/CH2-21 distance=4.07 Å). On the basis of the correlation between CH3-20 and CH2-21 observed in the 2D NOESY spectrum, the R-configuration of C-17 was concluded (see Supplementary Information).
A detailed literature search revealed that compound 3 was the first nucleoside derivative possessing imidazolidin-4-one framework. This prompted us to engage in the biosynthetic means of this exceptional ring system. Although the structure of amicetin, the originator of 3, was established in 1962, characterization of its biosynthesis was reported in 2012 by Zhang et al.16 In that study, first, the amicetin biosynthesis gene cluster from S. vinaceusdrappus NRRL 2363 was cloned, followed by heterologous expression in S. lividans TK64, which resulted in the production of amicetin and its analogs, confirming the identity of the gene cluster. Total of 21 genes, including 3 for regulation and transportation, 10 for disaccharide biosynthesis and 8 for the formation of the amicetin skeleton by the linkage of cytosine, p-amino benzoic acid and the terminal α-methylserine moieties, were described putatively in amicetin biosynthesis by in silico sequence analysis. In the case of 3, after biosynthesis of amicetin, an additional enzymatic step is required for a carbon transfer followed by a ring formation between N-15 and N-18 atoms. A comprehensive search pointed to N5,N10-methylene tetrahydrofolate (THF), an important metabolite for the synthesis of purines, thymidine, methionine, choline and lipids as the major source of C1 units in biosynthesis. Conversion of THF to 5,10-methylene-THF is one of the well-known enzymatic processes: the serine hydroxymethyltransferase, a pyridoxal 5′-phosphate-dependent enzyme, converts serine to glycine, transferring a methyl group to THF, thus producing 5,10-methylene-THF (Figure 3).26, 27, 28 With respect to the additional carbon’s incorporation (C-21), a plausible mechanism is proposed (Figure 4), which involves a serine hydroxymethyltransferase homolog in generation of the imidazolidine ring.27, 28, 29

SHMT catalyzed 5,10-methylene-THF formation. PLP, pyridoxal 5′-phosphate; SHMT, serine hydroxymethyltransferase; THF, tetrahydrofolate.

A plausible mechanism for the transformation of amicetin into compound 3. PLP, pyridoxal 5′-phosphate.
To the best of our knowledge, such a methylene insertion between the p-amino of p-amino benzoic acid moiety and the nitrogen atom of d-serine residue is encountered for the first time in secondary metabolites.
In addition to 3, another new metabolite (4) with a simple structural core was isolated. The HR-TOF-MS of 4 provided [M−H]− and [M+H]+ at m/z 245.0593 and 247.0864, respectively, indicating the molecular formula C11H10N4O3. Inspection of 1H and 13C NMR spectra of 4 suggested the presence of a 6-monosubstituted uracil residue instead of the cytosine moiety established in compounds 1–3 and 5.28, 29, 30 The substituted part was deduced to be p-amino benzamide as in the other isolates. The assignment of the 1H and 13C NMR signals of 4 was secured by COSY, HMQC and HMBC experiments (Table 1). On the basis of the spectral evidence, the structure of 4 was established as shown in Figure 1.
Amicetin-type nucleoside analogs are not common in nature, and about 10 derivatives have been identified so far, namely bamicetin,14 plicacetin,14 norplicacetin,15 oxamicetin,21 SF2457,31 oxyplicacetin32 and cytosaminomycins A to D.33 Amicetin and its analogs have been described as potent antibiotics against microorganisms, including archaea, bacteria and eukarya. These compounds exhibit their activity via peptidyl transferase inhibition blocking protein biosynthesis.34 Compounds 1–5 showed stronger activity than the positive controls against the test organisms with MIC values ranging from 4 to16 μg ml−1 (Table 2), substantiating antibiotic property of cytosine-type nucleosides towards microorganisms from different origins.
Materials and methods
Sampling station and isolation
A marine sediment sample, collected from Kaş, Turkey at a depth of 35 m by Scuba diving, was used for isolation of marine actinomycetes. The isolation procedure on the sediment was performed according to the method of Maldonado et al.35 M1 medium (10 g soluble starch, 4 g yeast extract, 2 g peptone and 15 g agar, 1000 ml seawater) was used for marine actinomycete isolation.36
Antimicrobial activity
Before the molecular characterization, a preliminary fermentation was performed with broth version of the same medium at 28 °C for 14 days at 150 r.p.m. to confirm antimicrobial activity. Sequential 25-ml aliquots of the fermentation broth were removed from the culture every 4 days from day 2 to 14, and activities of the ethyl acetate extracts were monitored using disc diffusion method on Mueller-Hinton agar against four different bacteria (Enteroccocus faecium DSM 13590: vancomycin resistant; E. coli O157:H7 RSKK 234: streptomycin, sulfisoxazole and tetracycline resistant; Staphylococcus aureus: methicillin resistant; Pseudomonas aureginosa ATCC 27853) and a yeast species (C. albicans DSM 5817).
Molecular identification
DNA isolation was carried out by a modified method of Liu et al.37 The isolate was identified using sequencing of the 16S ribosomal DNA region.38 Primers for amplification of the 16S ribosomal RNA region were 27F (5′-AGA GTT TGA TCC TGG CTC AG-3′) and 1492R (5′-TAC GGC TAC CTT GTT ACG ACT T-3′). The amplification reaction was performed with Corbett Cool Gradient Palm Cycler CGI-96 (Concorde, Sydney, NSW, Australia) under the following conditions as described elsewhere.13 Sequence analysis of 16S ribosomal RNA region was carried out on an ABI 3130XL automated sequencer (Applied Biosystems, Foster City, CA, USA). The sequence (JX912315.1) obtained was compared pairwise using a BLASTN search and aligned with the sequences of related species retrieved from GenBank, NCBI.
Large-scale fermentation
For large-scale fermentation, a loopful culture of S. rochei 06CM016 was inoculated in a flask containing 50 ml of M1 medium, and it was incubated at 28 °C for 3 days on a rotary shaker at 150 r.p.m. Five milliliter of the culture medium was transferred into 200 ml of M1 liquid medium in 1-l flasks, and further incubation was carried out at 28 °C for 10 days. In total, 56 l of fermentation liquid was obtained, and after removal of the cells by centrifugation, the broth was gradually extracted with EtOAc. After evaporation at 40 °C in vacuo, EtOAc extract was acquired (1.92 g; 0.034 g l−1 yield).
Thin-layer chromatography
To compare chemical compositions of fractions exhibiting activity and to determine the required solvent systems for chromatographical separations, silica gel (aluminum foil, prepared plate, Kieselgel 60 F254, 0.2 mm, Merck, Art.5554, Merck KGaA, Darmstadt, Germany) TLC analyses were performed. Solvent systems consisting of CHCl3:MeOH (9:1), CHCl3:MeOH:H2O (85:15:0.5, 80:20:2, 70:30:3 and 61:32:7), EtOAc:MeOH:H2O (100:17.5:13.5) and Hexane:EtOAc:MeOH (10:10:2 and 10:10:3) were used.
Bioassay-guided fractionation
Silica gel (Kieselgel 60, 70–230 mesh, Merck), reversed-phase silica gel (RP-C18, Sepralyte 40 μm) and Sephadex LH-20 as stationary phases on preparative thin layer, open column and high-performance flash chromatography (Biotage, Uppsala, Sweden) systems (see isolation scheme in the Supplementary Material) were used for purification studies.
Acidic hydrolysis of compound 1
Compound 3 (3.1 mg) was refluxed in 1 m HCl for 4 h. After removal of the solvent, the residue was partitioned between CHCl3 and H2O. The H2O solution was neutralized with 5% NaOH, and desalted on Sephadex LH-20 column by passing MeOH. The sugar fraction was concentrated in vacuo and purified on preparative TLC using CHCl3:Me2CO (1:1) to afford two saccharides, providing positive color reaction to anisaldehyde-H2SO4 (3a: 0.3 mg; 3b: 0.4 mg). Positive optical rotation data were observed for both sugar moieties (d-amicetose:  (c 0.0023, Me2CO); d-amosamine
(c 0.0023, Me2CO); d-amosamine  (c 0.0024, Me2CO)).
(c 0.0024, Me2CO)).
Broth microdilution method
MICs of the purified compounds were performed by the microdilution method described by CLSI.39
References
Bèrdy, J. Bioactive microbial metabolites. J. Antibiot. (Tokyo) 58, 1–26 (2005).
Lam, K. S. Discovery of novel metabolites from marine actinomycetes. Curr. Opin. Microbiol. 9, 245–251 (2006).
Sathiyaseelan, K. & Stella, D. Isolation, identification and antimicrobial activity of marine actinomycetes isolated from Parangipettai. Recent Res. Sci. Technol. 3, 74–77 (2011).
Thenmozhi, M. & Krishnan, K. Anti‐Aspergillus activity of Streptmyces sp. VITSTK7 isolated from bay of Bengal coast of Puducherry, India. J. Nat. Environ. Sci. 2, 1–8 (2011).
Koehn, F. E. & Carter, G. T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 4, 206–220 (2005).
Talbot, G. H. et al. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin. Infect. Dis. 42, 657–668 (2006).
Fenical, W. et al. in From Monsoons to Microbes: Understanding the Ocean’s Role in Human Health (ed Fenical, W.) 71–86 National Academies Press, Washington, (1999).
Alvarez-Mico, X., Jensen, P. R., Fenical, W. & Hughes, C. C. Chlorizidine, a cytotoxic 5H-pyrrolo[2,1-a]isoindol-5-one-containing alkaloid from a marine Streptomyces sp. Org. Lett. 15, 988–991 (2013).
Nam, S. J., Kauffman, C. A., Paul, L. A., Jensen, P. R. & Fenical, W. Actinoranone, a cytotoxic meroterpenoid of unprecedented structure from a marine adapted Streptomyces sp. Org. Lett. 15, 5400–5403 (2013).
Donia, M. & Hamann, M. T. Marine natural products and their potential applications as anti-infective agents. Lancet Infect. Dis. 3, 338–348 (2003).
Hughes, C. C., Prieto-Davo, A., Jensen, P. R. & Fenical, W. The marinopyrroles, antibiotics of an unprecedented structure class from a marine Streptomyces sp. Org. Lett. 10, 629–631 (2008).
Blunt, J. W., Copp, B. R., Munro, M. H. G., Northcote, P. T. & Prinsep, M. R. Marine natural products. Nat. Prod. Rep. 21, 1–49 (2004).
Özcan, K. et al. Diversity and antibiotic-producing potential of cultivable marine-derived actinomycetes from coastal sediments of Turkey. J. Soil. Sediment. 13, 1493–1501 (2013).
Haskell, T. H. et al. The isolation and characterization of three crystalline antibiotics from Streptomyces plicatus. J. Am. Chem. Soc. 80, 743–747 (1958).
Evans, J. R. & Weare, G. Norplicacetin, a new antibiotic from Streptomyces plicatus. J. Antibiot. 30, 604–606 (1977).
Zhang, G. et al. Characterization of the amicetin biosynthesis gene cluster from Streptomyces vinaceusdrappus NRRL 2363 implicates two alternative strategies for amide bond formation. Appl. Environ. Microbiol. 78, 2393–2401 (2012).
Latosińska, J. N., Seliger, J., Grechishkin, V. S. & Spychala, J. Studies of the electronic structure of 4-N-cytosine derivatives by NMR-NQR double resonance spectroscopy. Magn. Reson. Chem. 37, 881–884 (1999).
Sugimura, H. & Watanabe, K. Formal synthesis of cytosamine—A component of nucleoside antibiotics, the amicetin family. Synth. Commun. 31, 2313–2321 (2001).
Flynn, E. H., Hinman, J. W., Caron, E. L. & Woolf, D. O. The chemistry of Amicetin, a new antibiotic. J. Am. Chem. Soc. 75, 5867–5871 (1953).
Stevens, C. L., Nagarajan, K. & Haskell, T. H. The structure of amicetin. J. Org. Chem. 2, 2991–3005 (1962).
Konishi, M., Naruishi, M., Tsuno, T., Tsukiura, H. & Kawaguchi, H. Oxamicetin, a new antibiotic of bacterial origin. II. Structure of oxamicetin. J. Antibiot. (Tokyo) 26, 757–764 (1973).
Glaeske, K. W. & West, F. G. Chirality transfer from carbon to nitrogen to carbon via cyclic ammonium ylides. Org. Lett. 1, 31–34 (1999).
Katarzyńska, J. et al. 4-Methylpseudoproline derived from α-methylserine—synthesis and conformational studies. Org. Biomol. Chem. 10, 6705–6716 (2012).
Selambarom, J., Carre, F., Fruchier, A., Roque, J. P. & Pavia, A. A. Stereochemical study of 1, 3-N,X-heterocycles derived from alpha-aminoacids and formaldehyde. Structural evidence for the existence of the anomeric effect. Tetrahedron 58, 4439–4444 (2002).
Kleyer, D. L. & Koch, T. H. A photochemical ring contraction of an imino lactam. J. Org. Chem. 47, 3145–3148 (1982).
Matthews, R. G. & Drummond, J. T. Providing one-carbon units for biological methylations. Chem. Rev. 90, 1275–1290 (1990).
Schirch, V. & Szebenyi, D. M. E. Serine hydroxymethyltransferase revisited. Curr. Opin. Chem. Biol. 9, 482–487 (2005).
Bardagí, J. I. & Rossi, R. A. A novel approach to the synthesis of 6-substituted uracils in three-step, one-pot reactions. J. Org. Chem. 73, 4491–4495 (2008).
Angelaccio, S. Extremophilic SHMTs: From structure to biotechnology. BioMed Res. Int. 2013, 1–10 (2013).
Al-Turkistani, A. A., Al-Deeb, O. A., El-Brollosy, N. R. & El-Emam, A. A. Synthesis and antimicrobial activity of some novel 5-alkyl-6-substituted uracils and related derivatives. Molecules 16, 4764–4774 (2011).
Itoh, J. & Miyadoh, S. SF2457, a new antibiotic related to amicetin. J. Antibiot. (Tokyo) 45, 846–853 (1992).
Chen, Y., Zeeck, A., Chen, Z. & Zahner, H. Studies on metabolites produced by Streptomyces ramuloses Tue-34. II. The structural elucidation of oxyplicacetin, a new member of amicetin group. Kangshengsu 10, 285–295 (1985).
Haneda, K. et al. Cytosaminomycins, new anticoccidial agents produced by Streptomyces sp KO-8119. 1. Taxonomy, production, isolation and physicochemical and biological properties. J. Antibiot. (Tokyo) 47, 774–781 (1994).
Kirillov, S., Porse, B. T., Vester, B., Woolley, P. & Garrett, R. A. Movement of the 3'-end of tRNA through the peptidyl transferase centre and its inhibition by antibiotics. FEBS Lett. 406, 223–233 (1997).
Maldonado, L. A. et al. Diversity of cultivable actinobacteria in geographically widespread marine sediments. Antonie van Leeuwenhoek 87, 11–18 (2005).
Mincer, T. J., Jensen, P. R., Kauffman, C. A. & Fenical, W. Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments. Appl. Environ. Microbiol. 68, 5005–5011 (2002).
Liu, D., Coloe, S., Baird, R. & Pedersen, J. Rapid mini-preparation of fungal DNA for PCR. J. Clin. Microbiol. 38, 473 (2000).
Hall, V., Evans, T. L. & Duerden, B. I. Identification of actinomyces, propionibacteria, lactobacilli and bifidobacteria by amplified 16S rDNA restriction analysis. Anaerobe 7, 55–57 (2001).
Clinical and Laboratory Standards Institute CLSI. document M100-S17 (2007).
Acknowledgements
This work was supported by the Scientific and Technological Research Council of Turkey (TUBITAK, Project No: 109S361) and the Scientific Research Foundation of Ege University (10-FEN-012).
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Aksoy, S., Uzel, A. & Bedir, E. Cytosine-type nucleosides from marine-derived Streptomyces rochei 06CM016. J Antibiot 69, 51–56 (2016). https://doi.org/10.1038/ja.2015.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2015.72
This article is cited by
-
Purification and biological analysis of antimicrobial compound produced by an endophytic Streptomyces sp.
Scientific Reports (2023)
-
Antimicrobial compounds from marine actinomycetes
Archives of Pharmacal Research (2020)
-
Engineering nucleoside antibiotics toward the development of novel antimicrobial agents
The Journal of Antibiotics (2019)
-
Nature’s combinatorial biosynthesis and recently engineered production of nucleoside antibiotics in Streptomyces
World Journal of Microbiology and Biotechnology (2017)
