
Abstract
The candidate archaeal phylum ‘Aigarchaeota’ contains microorganisms from terrestrial and subsurface geothermal ecosystems. The phylogeny and metabolic potential of Aigarchaeota has been deduced from several recent single-cell amplified genomes; however, a detailed description of their metabolic potential and in situ transcriptional activity is absent. Here, we report a comprehensive metatranscriptome-based reconstruction of the in situ metabolism of Aigarchaeota in an oxic, hot spring filamentous ‘streamer’ community. Fluorescence in situ hybridization showed that these newly discovered Aigarchaeota are filamentous, which is consistent with the presence and transcription of an actin-encoding gene. Aigarchaeota filaments are intricately associated with other community members, which include both bacteria (for example, filamentous Thermocrinis spp.) and archaea. Metabolic reconstruction of genomic and metatranscriptomic data suggests that this aigarchaeon is an aerobic, chemoorganoheterotroph with autotrophic potential. A heme copper oxidase complex was identified in the environmental genome assembly and highly transcribed in situ. Potential electron donors include acetate, fatty acids, amino acids, sugars and aromatic compounds, which may originate from extracellular polymeric substances produced by other microorganisms shown to exist in close proximity and/or autochthonous dissolved organic carbon (OC). Transcripts related to genes specific to each of these potential electron donors were identified, indicating that this aigarchaeon likely utilizes several OC substrates. Characterized members of this lineage cannot synthesize heme, and other cofactors and vitamins de novo, which suggests auxotrophy. We propose the name Candidatus ‘Calditenuis aerorheumensis’ for this aigarchaeon, which describes its filamentous morphology and its primary electron acceptor, oxygen.
Similar content being viewed by others


Introduction
Members of the candidate archaeal phylum ‘Aigarchaeota’ were first identified in a subsurface hydrothermal ecosystem (Hirayama et al., 2005; Nunoura et al., 2005; Nunoura et al., 2011) and recently in a terrestrial hot spring as part of the Genome Encyclopedia of Bacteria and Archaea (GEBA) project utilizing single-cell genomics (Rinke et al., 2013). Aigarchaeota-related 16S rRNA genes have been detected in numerous terrestrial, subsurface, and marine hydrothermal environments (Hirayama et al., 2005; Nunoura et al., 2005; Costa et al., 2009; Vick et al., 2010; Meyer-Dombard et al., 2011; Cole et al., 2013; De Leon et al., 2013; Takacs-Vesbach et al., 2013). Phylogenetic placement of Aigarchaeota remains ambiguous, but recent phylogenomic studies reveal a close relationship with the Thaumarchaeota and possibly Crenarchaeota (Brochier-Armanet et al., 2011; Nunoura et al., 2011; Rayman et al., 2014). These reports have provided important insight into the phylogeny and evolution of the Aigarchaeota, yet little is known about the metabolic potential or in situ activity of members of this phylum. Near-complete genome sequences have been generated for several members of the Aigarchaeota using cultivation-independent methods including fosmids (Nunoura et al., 2011), single-cell amplified genomes (Rinke et al., 2013; Alba et al., 2014; Hedlund et al., 2014) and metagenomics (Alba et al., 2014; Hedlund et al., 2014). However, limited genome characterization and metabolic activity analysis has been completed on any members of this group (Nunoura et al., 2011; Rinke et al., 2013), especially those Aigarchaeota important in geothermal ecosystems of Yellowstone National Park (YNP). Moreover, no attempts have been made to link members of the Aigarchaeota with microbial activity (for example, metatranscriptomics) in relation to other community members and their physicochemical environment.
The environmental genome of Candidatus ‘Caldiarchaeum subterraneum’ was recovered from a mildly acidic (pH=5.1), oxic (~10 μm dissolved oxygen (DO)) subsurface filamentous ‘streamer’ community (Hirayama et al., 2005; depth ~320 m) at a temperature of 70 °C, and provided the first insight into the evolution and potential metabolism of members of the Aigarchaeota (Nunoura et al., 2011). Available genomic information from members of this deeply-rooted archaeal lineage suggests a possible evolutionary linkage with Eukarya (for example, an ubiquitin protein modification system; Nunoura et al., 2011). Populations of C. subterraneum might grow via the oxidation of hydrogen or carbon monoxide (CO) coupled to oxygen reduction by a heme copper (terminal) oxidase (HCO) complex. It is possible that these organisms can fix carbon dioxide via the 3-hydroxypropionate/4-hydroxybutyrate or dicarboxylate/4-hydroxybutyrate cycle (Nunoura et al., 2011). However, C. subterraneum lacked the key enzyme (4-hydroxybutyryl-CoA dehydratase) of these pathways (Berg et al., 2010, Nunoura et al., 2011). Genome sequence of two different Aigarchaeota single-cells were obtained from oxic (DO=42 μm), high temperature (81°C), circumneutral (pH=7.1) sediments at Great Boiling Spring (Rinke et al., 2013). Although the metabolic reconstruction of these Aigarchaeota was not a major focus of that study, the use of oxygen as an electron acceptor appears to be a consistent trait based on the presence of HCOs (Rinke et al., 2013; Alba et al., 2014; Hedlund et al., 2014). In addition, it has been speculated that some Aigarchaeota may also utilize other electron acceptors, such as oxidized sulfur or nitrogen compounds, but experimental data supporting these traits are unavailable (Nunoura et al., 2011; Alba et al., 2014; Hedlund et al., 2014).
The filamentous ‘pink streamer’ communities at Octopus Spring (OS) (Lower Geyser Basin, YNP) have been observed, studied, and intrigued scientists for more than 100 years (Setchell, 1903; Brock, 1967; Bauman and Simmonds, 1969; Stahl et al., 1985; Reysenbach et al., 1994; Jahnke et al., 2001; Blank et al., 2002). A recent study of the three predominant lineages of Aquificales in YNP (Takacs-Vesbach et al., 2013) showed that the high temperature (~82 °C) OS streamer community contained several uncharacterized and uncultured taxa, including a novel Aigarchaeota population. The presence of Aigarchaeota in OS presents an excellent opportunity to understand the metabolic capabilities and the activity of this population in situ. Consequently, the objectives of this study were to (i) obtain a detailed genome-based metabolic reconstruction of the predominant Aigarchaeota population present in OS, YNP, (ii) identify the morphology and spatial arrangement of Aigarchaeota and adjacent Thermocrinis spp. using fluorescence in situ hybridization (FISH), and (iii) determine the in situ transcriptional activity of this Aigarchaeota population to elucidate metabolic processes and potential community interactions.
Materials and methods
Site description and geochemistry
OS (Figure 1; Thermal Inventory ID LWCG138) is an alkaline siliceous geothermal spring located in the White Creek Area of the Lower Geyser Basin Region (lat 44.534083611, long −110.79788944) of YNP, WY, USA. The sample site discussed herein is classified as a ‘pink’ filamentous ‘streamer’ community (Figure 1), which has been the subject of many prior studies (for example, Bauman and Simmonds, 1969; Stahl et al., 1985; Reysenbach et al., 1994; Jahnke et al., 2001; Blank et al., 2002; Takacs-Vesbach et al., 2013). The metagenome sample discussed here was part of a 20-site study in YNP that included Aquificales-dominated ecosystems (Takacs-Vesbach et al., 2013; Inskeep et al., 2013). These streamer communities colonize habitats across a temperature range of 78–84 °C, pH~8 and DO ~40 μm (Supplementary Table S1). The streamer communities are considered oxic because the DO concentration is well above 0.2 μm, the (Km) of microbial HCO complexes (Morris and Schmidt, 2013) that were previously identified in the OS streamer community (Takacs-Vesbach et al., 2013). Microbial streamers present in the outflow channel exhibit a ‘streaming’ or oscillating motion in the high-velocity (~0.5 m s−1) outflow channel of OS (Supplementary Movie S1). Details on aqueous geochemical and physical sampling are discussed elsewhere (Inskeep et al., 2013; Takacs-Vesbach et al., 2013).

Photograph of a pink filamentous ‘streamer’ community found in the outflow channel of OS (24 July 2014).
DNA extraction and metagenome sequencing
Community DNA was extracted from OS ‘pink streamer’ biomass (Figure 1) sampled on 23 August 2007 by enzymatic and bead-beating lysis, pooled and sequenced using Sanger technology at the Department of Energy-Joint Genome Institute, Walnut Creek, CA, USA (see Takacs-Vesbach et al., 2013 for details on sequencing and assembly). In addition, Illumina sequencing was performed at Department of Energy-Joint Genome Institute to increase sequencing depth and coverage on two samples collected on 13 October 2011 and 5 October, 2012 from DNA extracted with the FastDNA Soil DNA extraction kit and protocol (MP Biomedicals LLC, Solon, OH, USA). The metagenomic sequence discussed here is publically available on the Integrated Microbial Genomes with Microbiome Samples under the IMG submission IDs 885, 10389 and 14458 for the 2007, 2011 and 2012 samples, respectively. Metagenome statistics from these samples are summarized in Supplementary Table S2.
De novo Aigarchaeota assembly analysis and annotation
Nucleotide word frequency-principal components analysis (NWF-PCA) of assembled metagenome sequence (Takacs-Vesbach et al., 2013) from OS revealed the presence of a novel Aigarchaeota lineage and several new bacterial taxa distantly related to the Thermotogae (Takacs-Vesbach et al., 2013; Colman et al., 2015). Other community members in OS include Thermocrinis spp., Pyrobaculum spp. and a novel Firmicutes population (Takacs-Vesbach et al., 2013; Jay et al., 2014; Coleman et al., 2015). A total of 1.16 Mb of sequence was recovered for the novel Aigarchaeota population with an average G+C content of 60.2% (Figure 2; Supplementary Table S3). The de novo aigarchaeon Sanger assembly (that is, Calditenuis aerorheumensis) was further analyzed with NWF-PCA (online web server, http://doug1dev.cgb.indiana.edu/uploadScatterplotForm3.html) and compared with other available Aigarchaeota sequence assemblies (>1 Mb), Caldiarchaeum subterraneum (Nunoura et al., 2011) and two single-cell amplified genomes from Great Boiling Spring (that is, aigarchaeota J15 and B22; Rinke et al., 2013). The word size was set to four, minimum FASTA size set to 5000 bp, normalize GC only, normalize values on and chop sequences to 5000 bp. Celera assembled sequence for the Aigarchaeota population from OS was annotated with Integrated Microbial Genome/Expert Review with Microbiome Samples (Markowitz et al., 2012) using Prodigal (Hyatt et al., 2010) to identify the open reading frames. Manual annotation of all genes and pathways was necessary because the annotation pipeline missed a number of archaeal central carbon pathways. Thus, BLASTp of the Calditenuis aerorheumensis de novo assembly with annotated genes of archaeal central carbon pathways was utilized (for example, Berg et al., 2007; Huber et al., 2008; Bräsen et al., 2014). Moreover, simple annotation of a gene based on a Cluster of Orthologous Gene assignment is misleading. For example, canonical aerobic CO dehydrogenases have been ‘identified’ in Aigarchaeota (Nunoura et al., 2011; Hedlund et al., 2014); however, this assignment is dubious given no evidence of necessary cofactor binding sites in the large subunit of this enzyme complex (Dobbek et al., 2002). Thus, it was necessary to align the deduced protein sequences of these genes (and others) with ClustalW (default alignment settings) to identify conserved cofactor binding and active sites with of known function and structure. The curated Calditenuis aerorheumensis assembly can be found on IMG/Mer under the name Aigarchaeota archaeon str. OS1 (Calditenuis aerorheumensis) and IMG submission ID 21516, and also under the NCBI BioProject ID PRJNA279181.

NWF-PCA (a) of the aigarchaeon de novo assembly (Calditenuis aerorheumensis, red) from OS compared with Candidatus ‘Caldiarchaeum subterraneum’ (yellow), and single-cell Aigarchaeota genomes from Great Boiling Spring (GBS): aigarchaeon B22 (blue) and aigarchaeon J15 (purple). The G+C content (%) of scaffolds (b) from C. aerorheumensis (red), GBS aigarchaeon B22 (blue) and GBS aigarchaeon J15 (purple) plotted as a function of decreasing scaffold length. The cumulative scaffold length is on the secondary y-axis (red, blue and purple lines represent C. aerorheumensi, GBS aigarchaeon B22 and GBS aigarchaeon J15, respectively). Deduced protein sequence comparison (c) of Calditenuis aerorheumensis versus aigarchaeon B22 (blue), aigarchaeon J15 (purple) and Caldiarchaeum subterraneum (yellow).
RNA extraction, sequencing and annotation
RNA was extracted from a matched sample of the OS ‘pink’ streamer community (13 October 2011) by modifying a FastRNA Pro-Soil-Direct Kit (MP Biomedicals) extraction kit and method. RNA from the streamer community was preserved in the field by placing streamer biomass directly into RNALater (Life Technologies, Carlsbad, CA, USA) and shaking vigorously for ~15 s. Streamer biomass (~0.5 g, n=3) was removed from the RNALater solution in a sterile, RNase-free hood, added to 0.5 ml of RNApro soil lysis solution in a lysing E matrix tube and vortexed for 15 min. The sample lysate was then centrifuged at 16 000 g for 10 min. The supernatant was transferred into a new 2 ml microcentrifuge tube (~500–800 μl), to which 1 ml of TriReagent was added and incubated for 5 min at room temperature (RT). Next, 200 μl of chloroform was added to the TriReagent/lysate mixture and incubated for 15 min at room temperature, followed by centrifugation at 16 000 g at 4 °C for 15 min. The triplicate supernatants containing RNA were pooled, and 500 μl of ice cold isopropanol (100%) was added and incubated overnight at −20 °C to facilitate RNA precipitation. RNA was pelleted by centrifugation at 16 000 g at 4 °C for 15 min and the pellet was washed once in ice cold 70% EtOH and centrifuged as before. The pellet was allowed to dry at room temperature for ~30 min, and immediately resuspended in 100 μl of TE buffer (pH=7, Life Technologies) and quantified with a Qubit 2.0 fluoremeter and Qubit RNA Broad Range Assay kit (Life Technologies). Total RNA quality was checked with a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). DNA contamination was analyzed by PCR with universal archaeal and bacterial 16S rRNA genes primers and ethidium bromide agarose gel electrophoresis. When DNA was present it was removed by DNase I treatment (New England Biolabs, Ipswich, MA, USA) for 30 min at 37 °C, followed by lithium chloride/EtOH precipitation overnight at −20 °C (Beam et al., 2014).
Double-stranded complementary DNA (cDNA) was synthesized at the Department of Energy-Joint Genome Institute from the OS total RNA extracts. In all, 2 μg of total RNA was subjected to rRNA depletion using Ribozero rRNA removal kit—Bacteria (Epicentre, Madison, WI, USA). Ribosomal RNA-depleted RNA was purified using Ampure XP beads (Agencourt, Beckman Coulter, Indianapolis, IN, USA), then fragmented using RNA Fragmentation Reagents (Life Technologies) at 70 °C for 2 min, targeting fragments ranging from 200 to 300 bp, then purified using Ampure XP beads. Reverse transcription was performed using SuperScript II Reverse Transcriptase (Life Technologies) with an initial annealing of random hexamers (Fermentas, Thermo Fisher Scientific, Pittsburgh, PA, USA) at 65 °C for 5 min, follow by an incubation of 42 °C for 50 min and an inactivation step at 70 °C for 10 min. Complementary DNA (cDNA) was then purified with Ampure XP beads, followed by second strand synthesis at 16 °C for 1 hour using a dNTP mix where dTTP is replaced by dUTP. Double-stranded cDNA fragments were purified and selected for targeted fragments (200–300 bp) using Ampure XP beads. The dscDNA was then blunt-ended, A-tailed and ligated with library adaptors using Kapa Library Amplification Kit (Kapa Biosystems, Wilmington, MA, USA) and purified using Ampure XP beads. Digestion of dUTP was then performed using AmpErase UNG (Applied Biosystems, Life Technologies) to remove second strand cDNA. Digested cDNA was purified with Ampure SPRI beads, followed by 10 cycles of PCR amplification using Kapa Library Amplification Kit. The final library was cleaned with Ampure SPRI beads and sequenced on an Illumina HiSeq 2000 platform generating paired-end reads of 150 bp.
Ribosomal RNA reads were removed in silico using the Kmer-based tool DUK (Li et al., 2011) and messenger RNA (mRNA)-enriched reads were mapped to the Calditenuis aerorheumensis assembly from OS utilizing Rockhopper, which uses a Bowtie2-like alignment algorithm (McClure et al., 2013). Mapped mRNA reads were standardized by the calculation reads per kilobase of transcript per million reads mapped (RPKM), which allows for the direct comparison of the level of transcription of all genes in the de novo assembly (Supplementary Table S4). RPKM is calculated in Rockhopper as: RPKM=((number of mapped reads/length of transcript (gene) in kilobase)/million mapped reads); McClure et al., 2013). Generally, if more reads map to a transcript of length x, and less map to another transcript with the same length x in the same sample, the former will have a higher RPKM value, and thus represent more in vivo transcriptional activity.
Phylogeny construction
A detailed 16S rRNA gene tree was constructed that contained a broad representation of members of the ‘Aigarchaeota’ in context with the Thaumarchaeota and Crenarchaeota. Aigarchaeota-related 16S rRNA genes were retrieved by BLAST homology from NCBI nr database including environmental sequences. Calditenuis aerorheumensis was also put into context by including 16S rRNA genes from recent Aigarchaeota single-cell genomes (Rinke et al., 2013; Alba et al., 2014; Hedlund et al., 2014). Sequences were aligned with ClustalW 1.6 implemented in MEGA 5.2.2 (Tamura et al., 2011) with default alignment settings. Alignments were manually inspected for obvious misaligned regions and introns, and removed from the final alignment (for example, most Thermoproteales contain 16S rRNA gene introns). A maximum-likelihood phylogenetic tree was created from the final alignment (1328 nucleotide positions) with the general time reversible model, estimated alpha parameter and four discrete gamma categories in MEGA 5.5.2 (100 bootstraps) (Tamura et al., 2011). Physical and aqueous geochemical data mapped onto the 16S rRNA gene tree is from Great Boiling Spring (Costa et al., 2009; Rinke et al., 2013), Little Hot Creek (Vick et al., 2010), Yellowstone Lake (Kan et al., 2011) and other YNP hot spring environments (Meyer-Dombard et al., 2011; Takacs-Vesbach et al., 2013).
A concatenated ribosomal protein phylogeny was constructed with 16 ribosomal proteins that were identified in all Aigarchaeota sequence assemblies (>1 Mb, n=4) shared with Thaumarchaeota, Crenarchaeota and Eukarya. The deduced proteins identified that fulfilled the above criteria were: L5, L10, L14, L15, L18, L19E, L24E, L24P, L44E, S6E, S8, S11P, S19, S27E and S28E. The deduced protein sequences were aligned individually with ClustalW 1.6 with default alignment parameters in MEGA 5.5.2 (Tamura et al., 2011). The resulting alignments were concatenated with a custom perl script (http://raven.iab.alaska.edu/~ntakebay/teaching/programming/perl-scripts/fastaConcat.pl). The concatenated aligned ribosomal proteins were manually inspected for regions where homology was doubtful and removed for final analysis. A maximum-likelihood phylogenetic tree was generated from the concatenated ribosomal proteins (2249 unambiguously aligned amino acids) using the WAG model and estimated alpha parameter (100 bootstraps) in MEGA 5.5.2 (Tamura et al., 2011).
FISH
Hot spring microbial filaments from OS (~82–84 °C) were collected on 28 August 2013 with a sterile scalpel and immediately fixed in 1% paraformaldehyde (final concentration) for 5 min at 4 °C. The fixative was then decanted and the sample was washed once in a 1:1 solution of 1 × phosphate-buffered saline:100% ethanol (EtOH) mixture (~10 ml) and stored in the 1:1 solution of 1 × phosphate-buffered saline: 100% EtOH at −20 °C until FISH. A FISH probe specific for all Group 1 A aigarchaeota (Figure 3; Aig800, Cy3-5′-GGC CCG TAG CCG CCC CGA CA-3′-Cy3; Supplementary Table S5) was manually designed from 16S rRNA gene alignments of this group around 16S rRNA sites accessible to probes (Behrens et al., 2003) and dual labeled with Cy3 (Integrated DNA Technologies, Coralsville, IA, USA) (Stoecker et al., 2010). Individual masses of microbial filaments were removed from the storage solution and rinsed three times in 0.22 μm filtered double-distilled water (ddH2O). Next, the ddH2O rinsed filaments were embedded in Tissue-Tek O.C.T (Sakura Finetek, Torrance, CA, USA) tissue media on a dry-ice block for cryosectioning and sectioned on a CM1800 cryostat (Leica Microsystems, Buffalo Grove, IL, USA). Thin sections were transferred to poly-lysine-coated glass microscope slides and stored at 4 °C until hybridization. Initial SYBR gold staining of thin sections revealed copious amounts of extracellular DNA present in the streamer community. Consequently, all thin sections utilized for FISH were pretreated with DNase I (New England Biolabs) for 30 min at 37 °C to degrade extracellular DNA, thus preventing non-specific probe binding. The DNase I reaction buffer was removed from the thin sections by rinsing in ddH2O (0.22 μm filtered) for ~1 min. Microscope slides containing thin sections (5 μm, three per slide) were dehydrated in an increasing EtOH series for 3 min each in 50, 80 and 100% EtOH, then air dried. Hybridization buffer (30 μl) containing 40% formamide, 0.9 M NaCl, 20 mm Tris HCL and 0.1% sodium dodecyl sulfate (added last to avoid precipitation) was added to each thin section and 1 μl of each probe (working solutions were 30 ng μl−1 for Cy3/Cy5 and 50 ng μl−1 for 6-FAM) to the buffer-coated thin sections. The slides were then placed in a 50 ml tube containing tissue paper soaked in the remaining hybridization buffer (~1 ml), sealed with the cap and placed in a 46 °C hybridization oven for 1.5 h. After hybridization, the slide was removed and immediately placed in 50 ml of pre-warmed wash buffer containing 46 mm NaCl, 20 mm Tris HCl and 5 mm EDTA for exactly 10 min in a 48 °C water bath. The slide was removed from the wash buffer, rinsed in ddH2O and dried with laboratory air. The slide was immediately visualized by confocal microscopy (Leica SP5 inverted confocal scanning laser microscope at the Confocal Microscope Facility at the Center for Biofilm Engineering, Montana State University, Bozeman, MT, USA) or stored at −20 °C for up to 3 days without loss of fluorescence. Non-specific probe binding to streamer thin sections was not observed using a probe that is not complementary to any known 16S rRNA (nonEub338). All FISH probes used in this study can be found in Supplementary Table S5.

FISH composite images (a, b) of OS filamentous streamer thin sections (5 μm) hybridized with the following DOPE probes: Cy3-Aig800 targeting Group 1A aigarchaeota (purple), Cy5-Arch915 (blue) and 6-FAM-Aqi338 (green). The aigarchaeota filaments are purple due to the overlapping emission from the Cy3 (red) and Cy5 (blue)-labeled probes. FISH composite image (c, d) with general probes Aqi338 (green), Eub338 (red) and Arch915 (blue). Scanning electron micrographs (e, f) of OS streamer community, showing filamentous organisms and condensed EPS and/or cellular appendages.
Scanning electron microscopy
Filamentous ‘pink streamer’ biomass was collected from OS on 12 October 2011 and fixed with 1% (final concentration) filter-sterilized glutaraldehyde. Streamer biomass was placed on a 0.22-μm filter and coated with iridium to reduce charging, and imaged with a Zeiss SUPRA 55VP field emission scanning electron microscope (Image and Chemical Analysis Laboratory, Montana State University) at low voltage (1 keV).
Results and Discussion
De novo Aigarchaeota assembly
The sequence assembly of the Aigarchaeota population from OS was compared with all available Aigarchaeota sequence assemblies (>1 Mb total sequence) using tetra-NWF-PCA (Figure 2a). The aigarchaeon from OS (Candidatus ‘Calditenuis aerorheumensis’, hereafter referred to as Calditenuis aerorheumensis) exhibits highly similar codon usage and G+C content to aigarchaeon B22 from Great Boiling Spring (GBS) (Figure 2a). Further G+C content analysis of assembled sequence confirmed the close relationship of C. aerorheumensis to aigarchaeon B22 with average G+C contents of 60.2 and 61.3%, respectively (Figure 2b). These two lineages have an average nucleotide identity of 87% among shared assembled sequence (~0.7 Mb), which suggests that they likely belong to the same novel candidate genus, Calditenuis. Calditenuis aerorheumensis and aigarchaeon B22 share a large amount of deduced protein sequences (~800) >80% amino-acid identity (Figure 2c), and also supports their inclusion in the same genus. Aigarchaeon J15 from GBS and Caldiarchaeum subterraneum each form separate clusters in NWF-PCA due to differences in codon usage bias and G+C content (Figure 2a and b). The relative abundance of C. aerorheumensis populations has been relatively stable over three sampling years at OS and represent 5.2±2.5% of all the random-sequence reads (Supplementary Table S1). A total of 1422 protein-encoding genes were predicted (Supplementary Table S3), which corresponds to a coding density of ~1.2 protein-encoding genes per kb genome, similar to the protein-encoding density in aigarchaeon B22 of 1.13 protein-encoding genes per kb genome. Genome completeness estimates from conserved archaeal single-copy genes (Rinke et al., 2013) suggest that the C. aerorheumensis assembly is ~80% complete, which equates to an estimated genome size of ~1.45 Mb. The high protein-coding density taken together with the small-predicted genome size suggests that this organism may have undergone extensive genome streamlining (Giovannoni et al., 2005, 2014).
FISH identification
All known Aigarchaeota encode for actin, which suggests that they are rods or filaments (Ettema et al., 2011); however, no definitive morphological observations have been made for any member of the Aigarchaeota. FISH with probe Aig800 designed to target terrestrial Group 1 A Aigarchaeota (including aigarchaeon B22) revealed that Calditenuis aerorheumensis populations in OS are filamentous (Figure 3a and b) ranging from 0.5 μm (diameter) by up to 20 μm (length), but may be longer due to damage during cryosectioning or sample dehydration. The broad-coverage archaeal FISH probe Arch915 also hybridized to these filaments (Figure 3a and b). Calditenuis aerorheumensis filaments were often found in close contact or vicinity of Thermocrinis spp. (Figure 3a and b), which were specifically detected using the newly designed probe Aqi338 (Kubo et al., 2011), and might be related to metabolite sharing and/or auxotrophic requirements of C. aerorheumensis. FISH probes targeting Thermocrinis spp., all bacteria, and all archaea revealed a close association of all microorganisms in the OS streamer community (Figure 3c and d). Archaea and Thermocrinis spp. are often found in helical bundled filaments, and Thermocrinis spp. can also be found in contact with other bacteria (Figure 3c and d). Scanning electron micrographs of the OS streamer community (Figure 3e and f) also confirm the filamentous habit of several population types, which range in diameters of ~0.2–0.5 μm up to ~0.7–1 μm. Small (<20 nm), extracellular polymeric substances (EPS) were evident in SEM images and represent a significant volume of the OS streamer community biomass (Figure 3e and f).
Phylogeny and distribution
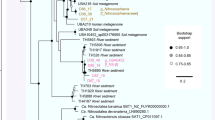
Phylogenetic analysis of 16S rRNA genes from the candidate phylum Aigarchaeota across numerous terrestrial, subsurface and freshwater geothermal environments revealed eight genus-level lineages (Figure 4). The C. aerorheumensis population groups with several YNP 16S rRNA gene clones from YNP Bison Pool ‘streamers’, as well as single-cell sequence (aigarchaeon B22) from GBS (Figure 4) at temperatures ranging from ~74 °C to 86 °C and pH values of 7.2–7.9. Group 1A Aigarchaeota are found in oxic hot spring ecosystems (DO ~10–53 μm), which is consistent across all the Aigarchaeota lineages currently identified by 16S rRNA genes. Aigarchaeota are distributed in geothermal environments at high temperature (68–87 °C) and moderately acidic to alkaline pH values (~5–9). The phylogenetic position of C. aerorheumensis was also confirmed using a conserved set of single-copy ribosomal proteins. The concatenated phylogenetic tree shows that C. aerorheumensis and aigarchaeon B22 are closely related, and form a separate group compared to C. subterraneum and aigarchaeon J15 (Figure 5).

Maximum likelihood 16S rRNA gene phylogenetic tree (1328 unambiguously aligned nucleotide positions) of the candidate phylum ‘Aigarchaeota’, Thaumarchaeota and Crenarchaeota rooted with Thermus thermophilus. Bootstrap (BS) values from 100 replications are included at the nodes; BS values <50% were not included. Scale bar represents the number of nucleotide substitutions per site.

Maximum likelihood phylogenetic tree of 16 concatenated ribosomal proteins (2249 amino-acid positions) from Calditenuis aerorheumensis compared with other Aigarchaeota, Thaumarchaeota, Crenarchaeota and Eukarya included as an outgroup. Numbers at the nodes represent bootstrap values from 100 replications and the scale bar represents the estimated amino-acid substitutions per site.
In situ metatranscriptome
The C. aerorheumensis assembly was utilized to align metatranscriptome reads from OS (~18 million mRNA enriched reads). A total of 544 290 mRNA reads were aligned to C. aerorheumensis, which constitutes ~3% of the total mRNA reads from the streamer community. Analysis of all mRNA reads using Cluster of Orthologous Genes showed that the transcription of a number of cellular processes were higher than their relative abundance in the genome (Supplementary Figure S1). Notably, energy production and conservation, and amino-acid transport and catabolism were over-transcribed compared with the relative abundance in the genome (Supplementary Figure S1). This finding suggests that amino-acid transport and catabolism is an important energy, carbon and nitrogen source for C. aerorheumensis in the streamer community.
Respiration
A detailed metabolic model was constructed for C. aerorheumensis using curated genome sequence and mRNA transcripts mapped to individual genes in each pathway (Figure 6). Pathways and metabolic functions that exhibited increased transcription levels were emphasized to reflect in situ activity (Figure 6). The streamer community present in OS (Figure 1) oscillates in the high-velocity channel (Supplementary Movie S1), which has aqueous O2 concentrations ranging from ~20 to 40 μm (Supplementary Table S1). Gradients in O2 are likely to occur within the streamer community that could create microaerobic or hypoxic microenvironments (for example, Bernstein et al., 2013). A single HCO complex (_00543–_00544) was identified in C. aerorheumensis that recruited numerous RNA reads from the metatranscriptome (RPKM ~70 000; Figure 6), and represents the dominant terminal electron acceptor utilized by C. aerorheumensis in OS. Similar HCO complexes were also identified in 60% of available Aigarchaeota sequence assemblies in IMG/Mer (Supplementary Table S7), which suggests that numerous members of this phylum are aerobic, and that O2 is an important niche-defining parameter for many members of this phylum. The importance of oxygen to the Aigarchaeota is also exemplified by the presence of genes that code for proteins responsible for degradation of reactive oxygen species in C. aerorheumensis, as well as 65% of Aigarchaeota genomes (Supplementary Table S7). An Fe-Mn superoxide dismutase was identified and transcription of this gene was high (RPKM ~800 000; Figure 6). A 2-Cys peroxiredoxin (_00775) and a 1-Cys peroxiredoxin (_00485) were identified and these enzymes are involved in the intra and extracellular degradation of hydrogen peroxide, respectively. Intracellular hydrogen peroxide is formed primarily by superoxide dismutase, and is hazardous to the cell by participating in Fenton reactions with Fe(II) that generate the DNA-damaging hydroxyl radical (•OH; Henle and Linn, 1997). Thus, the transcription of the 2-Cys peroxiredoxin is very high (RPKM ~130 000) and is comparable to the transcription levels of superoxide dismutase (RPKM ~800 000). Degradation of extracellular hydrogen peroxide by 1-Cys peroxiredoxin (RPKM ~15 000), which can form by the interaction of UV light and dissolved OC with O2 (Draper and Crosby, 1983), may be necessary for survival in these environments and/or an important ecosystem service that C. aerorheumensis performs for the OS streamer community, similar to Vibrio pelagius and Synechococcus spp. in the open ocean (Petasne and Zika, 1997).

Detailed metabolic reconstruction of Calditenuis aerorheumensis from OS based on environmental genome sequence and standardized mRNA reads (reported as RPKM; McClure et al., 2013). Numbers in colored boxes correspond to enzymes in specific pathways (Supplementary Table S6). ABC, adenosine triphosphate binding cassette; BC, blue copper protein (membrane bound); CO?, potential aerobic CO dehydrogenase complex; DOP, dissolved organic phosphorous; ED, Entner–Doudoroff; EMP, Embden–Meyerhof–Parnas glycolysis; GS, glyoxylate shunt/bypass; HCO, HCO complex (subunits I+III, II); II, succinate dehydrogenase complex; NUO, NADH dehydrogenase complex; PPP, oxidative pentose phosphate pathway; PRX, 1 and 2-cysteine peroxiredoxin; SOD, superoxide dismutase; TCA, tricarboxylic acid cycle; TTR, tetrathionate reductase complex.
The C. aerorheumensis population in OS also contains an operon encoding a putative tetrathionate reductase (TTR) system (_00420–_00424). To date, archaeal TTRs have only been identified in Pyrobaculum spp. and Archaeoglobus fulgidus (Cozen et al., 2009, Liebensteiner et al., 2013). The TTR operon was transcribed at low abundance (RPKM ~9000; Figure 6) and suggests that reduction of other terminal electron acceptors may provide an alternative to oxygen. Tetrathionate (S4O62−) has not been detected in OS; however, Thermocrinis spp. contain genes necessary for the oxidation of reduced sulfur compounds (Takacs-Vesbach et al., 2013) and may produce S4O62− as a byproduct, a feature that has been commonly observed in marine bacteria (Podgorsek and Imhoff, 1999).
Reducing equivalents (for example, NADH) generated from the oxidation of OC substrates (see below) are fed into the electron transport chain at the NADH dehydrogenase complex (Figure 6; complex I), which is not encoded in an operon. The membrane-bound succinate dehydrogenase complex (Figure 6; complex II) also feeds the electron transport chain. The C. aerorheumensis assembly does not encode a typical cytochrome C complex III for electron transport to complex IV (HCO); however, several membrane-bound blue copper proteins may serve as electron carries to the HCO or TTR complexes (Figure 6; _00503–_00505). The C. aerorheumensis population from OS has a complete electron transport chain, predominantly coupled to the reduction of oxygen, and adenosine triphosphate (ATP) synthesis driven by an archaeal V-type ATP synthase (Figure 6).
Energy metabolism
Calditenuis aerorheumensis contains genes necessary for the degradation of different organic carbon (OC) compounds coupled with adenosine triphosphate (organotrophy) and biomass (heterotrophy) synthesis. Major carbon substrates utilized for growth include acetate, fatty acids, amino acids and sugars. These OC constituents may originate from EPS produced by other thermophilic bacteria and archaea in OS and/or from autochthonous dissolved OC (Supplementary Table S1). Acetate is transported by a putative citrate/acetate antiporter (_00932; RPKM ~20 000), and utilized by an acetyl-CoA synthetase (_00897) to produce acetyl-CoA (Figure 6), which can then be fed into the tricarboxylic acid (TCA) cycle or gluconeogenesis. Acetate utilization is further supported by the presence of a functional glyoxylate bypass (Figure 6). The presence of isocitrate lyase (_00945; RPKM ~29 000) and malate synthase (_01089; RPKM ~70 000; Figure 6) provides a mechanism to incorporate carbon from acetate into biomass without the loss of CO2 through the TCA cycle. C. aerorheumensis also has the capacity to utilize fatty acids produced by thermophilic bacteria in OS as carbon and energy sources. This β-oxidation pathway contains an acyl-CoA synthetase (_01439; RPKM ~34 000) that converts fatty acids to acyl-CoA derivatives utilized by three different acyl-CoA dehydrogenases (_01040; _01032; _00259), which are likely specific for fatty acids of certain chain lengths (for example, odd or even). Transcription of one acyl-CoA dehydrogenase was considerably greater than several others (_01032; RPKM ~120 000) and likely corresponds to a preferred fatty acid utilized in situ by C. aerorheumensis. Although the metabolism of fatty acids appears to be distributed across most archaea (Dibrova et al., 2014), no enzymatic characterization of the different acyl-CoA dehydrogenases has been conducted. Physiological studies have shown that Archaeoglobus fulgidus can grow on either short- and/or long-chain fatty acids (Khelifi et al., 2010).
C. aerorheumensis also exhibits the metabolic capacity to utilize numerous amino acids and oligopeptides as carbon and energy sources. Transporters for amino acids were identified in the de novo assembly (Figure 6), and branched-chain amino acids are transported by a high-affinity ABC transporter that was highly transcribed (Figure 6; RPKM>100 000). A single copy of a dihydroxy acid dehydratase (ivlD; _01261) was identified, which may catalyze the conversion of branched-chain amino acids to TCA intermediates. Other amino acids are catabolized by a 2-oxoisovalerate oxidoreductase complex (_00164+_00741), which was also highly transcribed (RPKM>70 000), then incorporated into the TCA cycle (Figure 6).
A set of genes containing LigA and LigB homologs were identified in a single operon (_01094–_01099) potentially involved in the degradation of lignin or other aromatic compounds. Transcription levels in this pathway were increased for a hypothetical protein (_01096; RPKM ~200 000) and a ferredoxin ring-hydroxylating dioxygenase (_01095; RPKM ~70 000). The aromatic compound utilized by this pathway could not be identified, but these genes are also found in other Aigarchaeota (Hedlund et al., 2014). The source pool and outflow channel of OS contain significant concentrations of dissolved OC (~60 μm; Supplementary Table S1), which would be sufficient to support heterotrophic populations of C. aerorheumensis.
It has been suggested that some members of the Aigarchaeota are capable of using CO as an energy source (Nunoura et al., 2011; Rinke et al., 2013; Hedlund et al., 2014), but physiological evidence is lacking. The C. aerorheumensis assembly contains five copies of putative large-subunit aerobic CO dehydrogenases (coxL) (COG1529; _00394; _00730; _01206; _01316; 01360). However, all putative CoxL homologs from Aigarchaeota lack the conserved active site residues (VAYRCSFR) identified in canonical aerobic CO dehydrogenases (Supplementary Figure S2; Dobbek et al., 2002). The gene order of the middle and small subunits (_00406; _00407) is identical to the aerobic CO oxidizer, Oligotropha carboxidovorans, and both contain necessary cofactor binding sites (Dobbek et al., 1999). The transcription of coxSML genes were elevated (Figure 6; RPKM>150 000), so the function of these enzymes is important to determine and may aid in interpreting in situ physiology. Dissolved CO concentrations measured in OS are below detection (~10 nm) and CO concentrations in other geothermal waters range from ~30 nm to below detection levels (Kochetkova et al., 2011). Consequently, C. aerorheumensis populations are not likely utilizing CO as a principal electron donor given the low concentrations of CO relative to other electron donors such as reduced OC compounds (for example, dissolved OC ~60 μm; Supplementary Table S1).
Central carbon metabolism
C. aerorheumensis exhibits two routes for the conversion of glucose to pyruvate via either Embden–Meyerhof–Parnas glycolysis or Entner–Doudoroff pathways (Figure 6). A putative glucose ABC transporter operon was identified (_00266–_00269) and transcription was low except for the periplasmic subunit (_00269; Figure 6). Gluconeogenesis is also possible and the key enzyme fructose 1,6-bisphosphate aldolase/phosphatase is present, which has the conserved residues (GKDDP) implicated in catalysis (Say and Fuchs, 2010). All genes required for a complete TCA cycle were also identified with the exception of fumarase (Figure 6). Transcription of TCA cycle genes were high (RPKM >10 000), which is consistent with apparent primary sources of OC that C. aerorheumensis populations utilize in situ (that is, acetate, fatty acids and amino acids). A complete oxidative pentose phosphate pathway was identified in C. aerorheumensis with the exception of gluconolactonase, which is also absent in other thermophilic Thaumarchaeota (Spang et al., 2012; Beam et al., 2014).
Autotrophic Crenarchaeota and Thaumarchaeota fix inorganic carbon dioxide via the 3-hydroxypropionate/4-hydroxybutyrate (HP/HB) or dicarboxylate/4-hydroxybuyrate (DC/HB) cycles, which share a key marker enzyme, 4-hydroxybutyryl-CoA dehydratase (4-BUDH; Berg et al., 2010). A gene for a Type-2 4-BUDH (_01438) was identified in the C. aerorheumensis assembly (Figure 7; Supplementary Figure S3); however, Type-2 4-BUDHs have yet to be biochemically characterized, and all share a conserved His-292, but lack conserved cysteine residues required for (4Fe-4S) cluster binding (Berg et al., 2007). The transcription of the Type-2 4-BUDH was very high (RPKM>100 000) in C. aerorheumensis (Figure 7). No other function for this gene can be assigned other than the conversion of 4-hydroxybutyryl-CoA to crotonyl-CoA, which suggests that the Aigarchaeota Type-2 4-BUDHs may be functional in the HP/HB cycle. Moreover, C. aerorheumensis encodes for every gene of the HP/HB pathway, with the exception of NADPH-dependent malonate semialdehyde reductase and methylmalonyl-CoA epimerase (Figure 7; Supplementary Table S8). This is the first activity-based measurement that suggests members of the Aigarchaeota may fix carbon dioxide via the HP/HB pathway.

The 3-hydroxypropionate/4-hydroxybutyrate CO2 fixation cycle that is potentially operative in Calditenuis aerorheumensis (modified from Berg et al., 2007). mRNA reads from OS mapped to this pathway are reported as RPKM to take into account varying transcript lengths of the corresponding genes. Enzymes in the cycle: 1, acetyl-CoA carboxylase; 2, malonyl-CoA reductase; 3, malonate semialdehyde reductase; 4, 3-hydroxypropionyl-CoA synthetase; 5, 3-hydroxypropionyl-CoA dehydratase; 6, acryloyl-CoA reductase; 7, propionyl-CoA carboxylase; 8, methylmalonyl-CoA epimerase; 9, methylmalonyl-CoA mutase; 10, succinyl-CoA reductase; 11, succinate semialdehyde reductase; 12, 4-hydroxybutyryl-CoA synthetase; 13, 4-hydroxybutyryl-CoA dehydratase; 14, crotonyl-CoA hydratase; 15, 3-hydroxybutyryl-CoA dehydrogenase; 16, acetoacetyl-CoA β-ketothiolase. Note: 1, missing the biotin carboxylase subunit of bifunctional acetyl/propionyl-CoA carboxylase. Genes and corresponding RPKM values are presented in Supplementary Table S8.
Cofactor, vitamin and amino-acid biosynthesis
Pathways responsible for the synthesis of all amino acids, F420, pantothenate (B5), pyroxidine (B6) and cobalamin salvage were identified in C. aerorheumensis (Table 1). Coenzyme F420 is utilized in the glucose-6-phosphate dehydrogenase enzyme in the oxidative pentose phosphate pathway in C. aerorheumensis. This cofactor was also identified in the Thaumarchaeota, methanogenic archaea, and ‘Geoarchaeota’ (Kozubal et al., 2012; Spang et al., 2012). On the basis of the available sequence data, C. aerorheumensis cannot synthesize heme, thiamin, riboflavin, niacin and biotin; however, putative transporters were identified for these essential cofactors and vitamins (Table 1). The absence of vitamin and cofactor biosynthesis pathways appears common within this group of archaea (Supplementary Table S9); however, there are only a few complete genomes available for this candidate phylum. The absence of heme biosynthetic genes in C. aerorheumensis and single-cell genomes from the Aigarchaeota (Supplementary Table S9) is surprising because heme is the essential cofactor for oxygen binding in HCOs, and these genes were highly transcribed (Figure 6). The putative heme transporter was found at low transcriptional abundance (Figure 6), which might suggest strict heme recycling in vivo, as high intracellular heme concentrations are often hazardous and/or lethal (Anzaldi and Skaar, 2010). Low transcription levels of transporters for other cofactors and vitamins (Figure 6; except for thiamin and niacin) suggests conservative in vivo control of vitamin and cofactor recycling. Widespread auxotrophy in all known members of Aigarchaeota (Supplementary Table S9) suggest that they rely on other community members to supply them with essential compounds for growth and reproduction. The energetic cost of maintaining complete vitamin and cofactor biosynthesis pathways may have outweighed the cost of acquiring them from external sources (Giovannoni et al., 2005). Calditenuis aerorheumensis encodes the capacity for natural DNA competence (dprA) also present in some Thermoproteales (Siebers et al., 2011), and may be able to re-acquire complete cofactor and biosynthesis pathways under selection pressure.
Motility and attachment
Archaeal flagella are more similar to type IV secretion systems found in bacteria and have nothing in common with bacterial flagella other than the use for motion or taxis (Jarrell and Albers, 2012). All genes within an operon for an archaeal flagellum were found in C. aerorheumensis (_00011–_00018), and were transcribed at low levels (Figure 6; RPKM<1500). Flagellar synthesis may be more important during streamer formation and could explain the downregulation of these genes in this ‘mature’ community. A pilus system (_01412–_01414) was also identified in C. aerorheumensis. All pilus genes were highly transcribed (Figure 6; RPKM>150 000) and may be involved in cell–cell contact and association of C. aerorheumensis with other community members observed in FISH images (Figure 3a and b). The pilus system in C. aerorheumensis may be involved in the formation of EPS, which are abundant and represent a substantial fraction of the community biomass (Figure 3e and f). Pili have been identified in other archaea as well, and are involved in cell–cell adherence and/or biofilm formation (Albers and Meyer, 2011).
Etymology
The provisional taxonomic assignment for this group of Aigarchaeota is ‘Candidatus: Calditenuis aerorheumensis’ alluding to its thermal habitat, filamentous shape and aerobic catabolism.
Calditenuis gen. nov.
Calditenuis aerorheumensis sp. nov.
Genus: Caldi (L. adj.): warm, tenuis (L. adj.): thin or slender. Species: aero (Gr. noun): air or atmosphere, rheumensis (Gr. noun); a stream, current, or that which flows. The Genus name describes the organism’s thermophilic nature and thin, filamentous shape. The species name alludes to the organism’s principal terminal electron acceptor, oxygen and filamentous ‘streamer’ habitat.
Locality: oxic (~30 μm), high temperature (~75–85 °C) and pH ~7.5–8.5 thermal springs.
Diagnosis: a thin, filamentous, aerobic chemoorganohetero(auto)troph from the Aigarchaeota.
Comment: Future efforts to isolate members of this genus and all other Aigarchaeota (with the exception of aigarchaeon J15) might consider adding heme to vitamin solutions, which is not common in Wolfe’s vitamin solution (Wolin et al., 1963) typically used in archaeal enrichment cultures. Moreover, C. aerorheumensis is likely oligotrophic and may not respond to high concentrations of C sources and/or vitamins.
References
Alba TW, Murugapiran SK, Blainey PC, Dodsworth JA, Thomas S, Woyke T et al. (2014). Insights into the global diversity and physiology of 'Aigarchaeota' through synergistic analysis of single-cell genomes and metagenomes. Ninth Annual DOE Joint Genome Institute User Meeting. Walnut Creek, CA, USA.
Albers S-V, Meyer BH . (2011). The archaeal cell envelope. Nat Rev Microbiol 9: 414–426.
Anzaldi L, Skaar E . (2010). Overcoming the heme paradox: heme toxicity and tolerance in bacterial pathogens. Infect Immun 78: 4977–4989.
Bauman AJ, Simmonds PG . (1969). Fatty acids and polar lipids of extremely thermophilic bacterial masses from two Yellowstone hot springs. J Bacteriol 98: 531.
Beam JP, Jay ZJ, Kozubal MA, Inskeep WP . (2014). Niche specialization of novel Thaumarchaeota to oxic and hypoxic acidic geothermal springs of Yellowstone National Park. ISME J 8: 938–951.
Behrens S, Rühland C, Inácio J, Huber H, Fonseca Á, Spencer-Martins I et al. (2003). In situ accessibility of small-subunit rRNA of members of the domains Bacteria, Archaea, and Eucarya to Cy3-labeled oligonucleotide probes. Appl Environ Microbiol 69: 1748–1758.
Berg I, Kockelkorn D, Buckel W, Fuchs G . (2007). A 3-hydroxypropionate/4-hydroxybutyrate autotrophic carbon dioxide assimilation pathway in archaea. Science 318: 1782–1786.
Berg I, Kockelkorn D, Ramos-Vera W, Say R, Zarzycki J, Hugler M et al. (2010). Autotrophic carbon fixation in archaea. Nature Rev Microbiol 8: 447–460.
Bernstein HC, Beam JP, Kozubal MA, Carlson RP, Inskeep WP . (2013). In situ analysis of oxygen consumption and diffusive transport in high-temperature acidic iron-oxide microbial mats. Environ Microbiol 15: 2360–2370.
Blank CE, Cady SL, Pace NR . (2002). Microbial composition of near-boiling silica-depositing thermal springs throughout Yellowstone national park. Appl Environ Microbiol 68: 5123–5135.
Bräsen C, Esser D, Rauch B, Siebers B . (2014). Carbohydrate metabolism in Archaea: current insights into unusual enzymes and pathways and their regulation. Microbiol Mol Biol Rev 78: 89–175.
Brochier-Armanet C, Forterre P, Gribaldo S . (2011). Phylogeny and evolution of the Archaea: one hundred genomes later. Curr Opin Microbiol 14: 274–281.
Brock TD . (1967). Life at high temperatures. Science 158: 1012–1019.
Cole JK, Peacock JP, Dodsworth JA, Williams JA, Thompson DB, Dong H et al. (2013). Sediment microbial communities in Great Boiling Spring are controlled by temperature and distinct from water communities. ISME J 7: 718–729.
Colman DR, Jay ZJ, Inskeep WP, Rusch DB, Jennings RdeM, Maas KR et al. (2015). Characterization of novel, deeply-branching bacterial populations recovered from YNP thermal metagenomes. ISME J (in review).
Costa KC, Navarro JB, Shock EL, Zhang CL, Soukup D, Hedlund BP . (2009). Microbiology and geochemistry of great boiling and mud hot springs in the United States Great Basin. Extremophiles 13: 447–459.
Cozen A, Weirauch M, Pollard K, Bernick D, Stuart J, Lowe T . (2009). Transcriptional map of respiratory versatility in the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. J Bacteriol 191: 782–794.
De Leon KB, Gerlach R, Peyton BM, Fields MW . (2013). Archaeal and bacterial communities in three alkaline hot springs in Heart Lake Geyser Basin, Yellowstone National Park. Front Microbiol 4: 330.
Dibrova D, Galperin M, Mulkidjanian A . (2014). Phylogenomic reconstruction of archaeal fatty acid metabolism. Environ Microbiol 16: 907–918.
Dobbek H, Gremer L, Meyer O, Huber H . (1999). Crystal structure and mechanism of CO dehydrogenase, a molybdo iron-sulfur flavoprotein containing S-selanylcysteine. Proc Natl Acad Sci USA 96: 8884–8889.
Dobbek H, Gremer L, Kiefersauer R, Huber R, Meyer O . (2002). Catalysis at a dinuclear [CuSMo(==O)OH] cluster in a CO dehydrogenase resolved at 1.1-Å resolution. Proc Natl Acad Sci USA 99: 15971–15976.
Draper W, Crosby D . (1983). The photochemical generation of hydrogen-peroxide in natural-waters. Arch Environ Contam Toxicol 12: 121–126.
Ettema T, Lindas A, Bernander R . (2011). An actin-based cytoskeleton in archaea. Mol Microbiol 80: 1052–1061.
Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vargin KL, Baptista D et al. (2005). Genome streamlining in a cosmopolitan oceanic bacterium. Science 309: 1242–1245.
Giovannoni SJ, Thrash JC, Temperton B . (2014). Implications of streamlining theory for microbial ecology. ISME J 8: 1553–1565.
Hedlund BP, Dodsworth JA, Murugapiran SK, Rinke C, Woyke T . (2014). Impact of single-cell genomics and metagenomics on the emerging view of extremophile “microbial dark matter”. Extremophiles 5: 865–875.
Henle ES, Linn S . (1997). Formation, prevention, and repair of DNA damage by iron/hydrogen peroxide. J Biol Chem 272: 19095–19098.
Hirayama H, Takai K, Inagaki F, Yamato Y, Suzuki M, Nealson K et al. (2005). Bacterial community shift along a subsurface geothermal water stream in a Japanese gold mine. Extremophiles 9: 169–184.
Huber H, Gallenberger M, Jahn U, Eylert E, Berg I, Kockelkorn D et al. (2008). A dicarboxylate/4-hydroxybutyrate autotrophic carbon assimilation cycle in the hyperthermophilic Archaeum Ignicoccus hospitalis. Proc Natl Acad Sci USA 105: 7851–7856.
Hyatt D, Chen G, LoCascio P, Land M, Larimer F, Hauser L . (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformat 11: 119.
Inskeep WP, Jay Z J, Tringe SG, Herrgard M, Rusch D B et al. (2013). The YNP Metagenome Project: environmental parameters responsible for microbial distribution in the Yellowstone geothermal ecosystem. Front Microbiol 4: 67.
Jahnke LL, Eder W, Huber R, Hope JM, Hinrichs K-U, Hayes JM et al. (2001). Signature lipids and stable carbon isotope analyses of Octopus Spring hyperthermophilic communities compared with those of aquificales representatives. Appl Environ Microbiol 67: 5179–5189.
Jarrell K, Albers S . (2012). The archaellum: an old motility structure with a new name. Trends Microbiol 20: 307–312.
Jay ZJ, Rusch DB, Romine MF, Beam JP, Inskeep WP . (2014). Geochemical constraints on the distribution and function of thermoproteales populations in Yellowstone national park. American Geophysical Union Conference. San Francisco, CA, USA.
Kan J, Clingenpeel S, Macur RE, Inskeep WP, Lovalvo S, Varley J et al. (2011). Archaea in Yellowstone Lake. ISME J 5: 1784–1795.
Khelifi N, Grossi V, Hamdi M, Dolla A, Tholozan J, Ollivier B et al. (2010). Anaerobic oxidation of fatty acids and alkenes by the hyperthermophilic sulfate-reducing archaeon Archaeoglobus fulgidus. Appl Environ Microbiol 76: 3057–3060.
Kochetkova T, Rusanov I, Pimenov N, Kolganova T, Lebedinsky A, Bonch-Osmolovskaya E et al. (2011). Anaerobic transformation of carbon monoxide by microbial communities of Kamchatka hot springs. Extremophiles 15: 319–325.
Kozubal MA, Romine M, Jennings RdeM, Jay ZJ, Tringe SG, Rusch DB et al. (2012). Geoarchaeota: a new candidate phylum in the Archaea from high-temperature acidic iron mats in Yellowstone National Park. ISME J 7: 622–634.
Kubo K, Knittel K, Amann R, Fukui M, Matsuura K . (2011). Sulfur-metabolizing bacterial populations in microbial mats of the Nakabusa hot spring, Japan. Syst Appl Microbiol 34: 293–302.
Li M, Copeland A, Han J . (2011). DUK - A fast ad Efficient Kmer Based Sequence Matching Tool. Lawrence Berkeley National Laboratory. LBNL Paper LBNL-4516E-Poster p.
Liebensteiner M, Pinkse M, Schaap P, Stams A, Lomans B . (2013). Archaeal (Per)Chlorate reduction at high temperature: an interplay of biotic and abiotic reactions. Science 340: 85–87.
Markowitz V, Chen I-M, Palaniappan K, Chu K, Szeto E, Grechkin Y et al. (2012). IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res 40: D115–D122.
McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco C et al. (2013). Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res 41: e140.
Meyer-Dombard DR, Swingley W, Raymond J, Havig J, Shock EL, Summons RE . (2011). Hydrothermal ecotones and streamer biofilm communities in the Lower Geyser Basin, Yellowstone National Park. Environ Microbiol 13: 2216–2231.
Morris R, Schmidt T . (2013). Shallow breathing: bacterial life at low O2 . Nat Rev Microbiol 11: 205–212.
Nunoura T, Hirayama H, Takami H, Oida H, Nishi S, Shimamura S et al. (2005). Genetic and functional properties of uncultivated thermophilic crenarchaeotes from a subsurface gold mine as revealed by analysis of genome fragments. Environ Microbiol 7: 1967–1984.
Nunoura T, Takaki Y, Kakuta J, Nishi S, Sugahara J, Kazama H et al. (2011). Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group. Nucleic Acids Res 39: 3204–3223.
Petasne R, Zika R . (1997). Hydrogen peroxide lifetimes in south Florida coastal and offshore waters. Mar Chem 56: 215–225.
Podgorsek L, Imhoff JF . (1999). Tetrathionate production by sulfur oxidizing bacteria and the role of tetrathionate in the sulfur cycle of Baltic Sea sediments. Aquat Microb Ecol 17: 255–265.
Rayman K, Forterre P, Brochier-Armanet C, Gribaldo S . (2014). Global phylogenomic analysis disentangles the complex evolutionary history of DNA replication in archaea. Genome Biol Evol 6: 192–212.
Reysenbach A-L, Wickham GS, Pace NR . (1994). Phylogenetic analysis of the hyperthermophilic pink filament community in Octopus Spring, Yellowstone National Park. Appl Environ Microbiol 60: 2113–2119.
Rinke C, Schwientek P, Sczyrba A, Ivanova N, Anderson I, Cheng J et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437.
Say RF, Fuchs G . (2010). Fructose 1,6-bisphosphate aldolase/phosphatase may be an ancestral gluconeogenic enzyme. Nature 464: 1077–1081.
Siebers B, Zaparty M, Raddatz G, Tjaden B, Albers S-V, Bell SD et al. (2011). The complete genome sequence of Thermoproteus tenax: a physiologically versatile member of the Crenarchaeota. PLoS One 6: e24222.
Setchell WA . (1903). The upper temperature limits of life. Science 17: 934–937.
Spang A, Poehlein A, Offre P, Zumbrägel S, Haider S, Rychlik N et al. (2012). The genome of the ammonia-oxidizing Candidatus Nitrososphaera gargensis: insights into metabolic versatility and environmental adaptations. Environ Microbiol 14: 3122–3145.
Stoecker K, Dorninger C, Daims H, Wagner M . (2010). Double labeling of oligonucleotide probes for fluorescence in situ hybridization (DOPE-FISH) improves signal intensity and increases rRNA accessibility. Appl Environ Microbiol 76: 922–926.
Stahl DA, Lane DJ, Olsen GJ, Pace NR . (1985). Characterization of a Yellowstone hot spring microbial community by 5S rRNA seqences. Appl Environ Microbiol 49: 1379–1384.
Takacs-Vesbach C, Inskeep W, Jay Z, Herrgard M, Rusch D, Tringe S et al. (2013). Metagenome sequence analysis of filamentous microbial communities obtained from geochemically distinct geothermal channels reveals specialization of three Aquificales lineages. Front Microbiol 4: 84.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S . (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739.
Vick TJ, Dodsworth JA, Costa KC, Shock EL, Hedlund BP . (2010). Microbiology and geochemistry of Little Hot Creek, a hot spring environment in the Long Valley Caldera. Geobiology 8: 140–154.
Wolin E, Wolin M, Wolfe R . (1963). Formation of methane by bacterial extracts. J Biol Chem 238: 2882–2886.
Acknowledgements
This work was supported by the Department of Energy (DOE)—Pacific Northwest National Laboratory Foundational Science Focus Area in Biological Interactions (Subcontract 112443), the DOE—Joint Genome Institute Community Sequencing Projects (CSP 787081 and 787701), the National Science Foundation—Integrative Graduate Education and Traineeship Program (JPB, ZJJ and R de MJ; DGE 0654336), and an Advanced Grant of the European Research Council to MW (MCS and MW; NITRICARE 294343). The work conducted by the Department of Energy-Joint Genome Institute, a DOE Office of Science User Facility, was supported by the Office of Science of the US DOE (contract no. DE-AC02-05CH11231). We appreciate Brian Hedlund and Tim Alba for data sharing, Christie Hendrix and Stacey Gunther (YNP Center for Resources) for research permitting in YNP (permits YELL-SCI-5068 and -5686) and Betsey Pitts for cryosection instruction and helpful comments on confocal microscopy. FISH images were taken at microscope facilities at the Center for Biofilm Engineering (Montana State University) funded by the NSF-MRI Program and the M.J. Murdock Charitable Trust. The open access fees were generously provided by the Library's Author Fund at Montana State University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Beam, J., Jay, Z., Schmid, M. et al. Ecophysiology of an uncultivated lineage of Aigarchaeota from an oxic, hot spring filamentous ‘streamer’ community. ISME J 10, 210–224 (2016). https://doi.org/10.1038/ismej.2015.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.83
This article is cited by
-
Microbial diversity in extreme environments
Nature Reviews Microbiology (2022)
-
Longitudinal analysis of the Five Sisters hot springs in Yellowstone National Park reveals a dynamic thermoalkaline environment
Scientific Reports (2022)
-
Culexarchaeia, a novel archaeal class of anaerobic generalists inhabiting geothermal environments
ISME Communications (2022)
-
An essential role for tungsten in the ecology and evolution of a previously uncultivated lineage of anaerobic, thermophilic Archaea
Nature Communications (2022)
-
Diversity, ecology and evolution of Archaea
Nature Microbiology (2020)
