
Abstract
Carbon fixation by chemoautotrophic microbes such as homoacetogens had a major impact on the transition from the inorganic to the organic world. Recent reports have shown the presence of genes for key enzymes associated with the Wood–Ljungdahl pathway (WLP) in the phylum Actinobacteria, which adds to the diversity of potential autotrophs. Here, we compiled 42 actinobacterial metagenome-assembled genomes (MAGs) from new and existing metagenomic datasets and propose three novel classes, Ca. Aquicultoria, Ca. Geothermincolia and Ca. Humimicrobiia. Most members of these classes contain genes coding for acetogenesis through the WLP, as well as a variety of hydrogenases (NiFe groups 1a and 3b–3d; FeFe group C; NiFe group 4-related hydrogenases). We show that the three classes acquired the hydrogenases independently, yet the carbon monoxide dehydrogenase/acetyl-CoA synthase complex (CODH/ACS) was apparently present in their last common ancestor and was inherited vertically. Furthermore, the Actinobacteria likely donated genes for CODH/ACS to multiple lineages within Nitrospirae, Deltaproteobacteria (Desulfobacterota), and Thermodesulfobacteria through multiple horizontal gene transfer events. Finally, we show the apparent growth of Ca. Geothermincolia and H2-dependent acetate production in hot spring enrichment cultures with or without the methanogenesis inhibitor 2-bromoethanesulfonate, which is consistent with the proposed homoacetogenic metabolism.
Similar content being viewed by others

Introduction
The Wood–Ljungdahl pathway (WLP) exists in a diversity of bacterial and archaeal lineages, and is considered one of the most ancient carbon fixation pathways [1]. The WLP can be used for either homoacetogenesis or methanogenesis in different taxa, and operates by reduction of CO2 to acetyl-CoA or in the reverse direction to oxidize acetate to H2 and CO2 under anoxic conditions [1,2,3,4]. In homoacetogens, two molecules of CO2 are reduced via hydrogen in two separate branches: the methyl branch, using tetrahydrofolate as a C1 carrier, and the carbonyl branch, using carbon monoxide dehydrogenase (CODH). Methyl and carbonyl groups from the two branches are then combined by acetyl-CoA synthase (ACS) to form acetyl-CoA, which can be used anabolically or converted to acetate [5]. Acetogens are phylogenetically diverse, belonging to 23 different genera, many of which belong to the Firmicutes [6]. In methanogens, the carbonyl branch is similar, but the methyl branch uses methanofuran and tetrahydromethanopterin as C1 carriers, and yields both methane and acetyl-CoA [7]. Several methanogens, such as Methanoregula boonei, Methanoregula formicica, and Methanocella arvoryzae, use the WLP only catabolically and do not fix CO2 [1].
The WLP is inextricably associated with acetogens and methanogens [1,2,3,4], but recently the genes for CODH/ACS were detected in metagenome-assembled genomes (MAGs) of Actinobacteria [8, 9], which was surprising given the heterotrophic lifestyle of cultivated Actinobacteria. A recent study has given the first insight into the catabolic function (from acetyl-CoA to H2 and CO2) of actinobacterial WLP; however, so far, there is no evidence that Actinobacteria can fix CO2 via the WLP, and the diversity and ecological function of Actinobacteria with the WLP is poorly explored [8, 9]. In this study, we aimed to uncover the distribution of Actinobacteria with the genomic potential for the WLP, investigate the evolutionary history of the WLP in this phylum, and provide genomic and experimental evidence of homoacetogenesis in Actinobacteria.
Materials and methods
Sample collection, DNA extraction, and metagenomic sequencing
A total of 11 hot spring sediment samples were collected by using sterile spatulas and spoons from Yunnan and Tibet, China (Fig. 1a; Table S1). Samples were immediately frozen in liquid nitrogen and stored at −80 °C before DNA extraction. Temperature, pH (measured in situ), and GPS coordinates of the sample locations are shown in (Table S1). These measurements and other geochemical analyses were performed as described earlier [10]. DNA from each sample was extracted from ~20 g of sediment by using the Powersoil DNA Isolation Kit (MoBio). A Qubit Fluorometer was used to measure the concentration of the extracted DNAs. Approximately 30 Gbp (2 × 150 bp) of metagenomic data for each sample was generated by using the Illumina HiSeq 4000 Platform using a 350 bp insert library at Beijing Novogene Bioinformatics Technology Co., Ltd (Beijing, China).

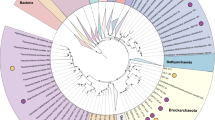
a Geographic locations of hot spring samples. b Phylogenetic placement of actinobacterial MAGs. Multiple sequence alignments of 120 marker genes generated by GTDB-Tk were used to construct the phylogenetic tree of Actinobacteria by IQ-Tree with the model LG+F+R10. Bootstrap values were based on 1000 replicates and nodes with percentages > 80% are indicated as black circles. Red stars illustrate the genomes recovered from hot spring sediments, and black stars represent the WLP-encoding MAGs reported in previous studies [8, 9]. All the MAGs belonging to Ca. Geothermincolia, Ca. Humimicrobiia, and Ca. Aquicultoria contain the genes coding for CODH/ACS, except RBG_19FT_COMBO_54_7 and UBA1414, marked by black rectangles. Outer circles are colored by environmental sources (see sources legend). c Relative abundance of MAGs. The relative abundance of each bin was calculated based on the number of reads mapped to each bin/the total number of reads.
Metagenome assembly and genome binning
The raw reads were quality filtered as described earlier [11]. Briefly, four steps were performed to obtain the clean reads: (1) adapter-contaminated reads were eliminated; (2) duplicated reads generated by PCR amplification were deleted; (3) reads with a significant excess of “N” (≥ 10% of the read) were removed; and (4) reads with quality score less than 15 at the 3′ end were trimmed. The high-quality reads of from each sample were de novo assembled using SPAdes (version 3.9.0) [12] individually with the following parameters: -k 33, 55, 77, 99, 111 --meta. Reads were mapped to scaffolds using BBMap (version 38.85; http://sourceforge.net/projects/bbmap/). Genome binning was conducted on scaffolds with length > 2.5 kbp using MetaBAT [13]. Completeness and contamination of each genome bin were calculated using CheckM [14]. A genomic database was established from previously described MAGs with genes for the WLP [8], and used to search for related groups in our hot spring metagenomic dataset (55 hot spring metagenomes) and public databases (99 MAGs from NCBI) by Genome Taxonomy Database (GTDB)-Tk [15] (de_novo_wf --outgroup_taxon p__Chloroflexota --taxa_filter p__Actinobacteria --bac120_ms). Finally, a total of 42 MAGs (15 MAGs from metagenomic dataset of hot spring and 27 MAGs from NCBI) belonging to three novel classes of Actinobacteria were identified.
Functional annotation of genomes
Putative protein-coding sequences (CDSs) were predicted using Prodigal [16] with the “-p single” parameter. The predicted CDSs were then annotated against KEGG, eggNOG, and NCBI-nr databases using DIAMOND [17] by applying E-values < 1e−5. For pathway analysis, the predicted CDSs were uploaded to KEGG Automatic Annotation Server [18] with “for prokaryotes” and “bidirectional best hit” options. The tRNAs and rRNAs were identified by tRNAscan-SE version 2.0.2 [19] and RNAmmer version 1.2, respectively [20].
Phylogenetic analysis
A reference actinobacterial genomic dataset for phylogenetic analysis was established by downloading type species genomes from NCBI and IMG-M databases (Table S2). To investigate whether members of Coriobacteriia contain the WLP, almost all the genomes of Coriobacteriia species (100 genomes) were included in our analysis. Genome quality was evaluated using CheckM [14] and genomes with estimated completeness < 80% or contamination > 5% were discarded from the study. Due to the lack of representatives in Candidatus (Ca.) Aquicultoria, MAGs from this class were included for the later analysis if their estimated completeness was >75%. The actinobacterial phylogeny was built from the multiple sequence alignments (MSAs) generated by GTDB-Tk software [15]. A maximum-likelihood phylogeny for MSAs was calculated by IQ-Tree [21] with parameters (-alrt 1000 -bb 1000 -nt AUTO). The best-fit model (LG+F+R10) determined by ModelFinder [22] is well supported by Akaike information criterion (AIC), corrected AIC (AICc), and Bayesian information criterion.
For phylogenetic analysis based on genes of interest, datasets were derived as described in the relevant literature [1, 8, 23,24,25]. Reference amino acid sequences for CODH/ACS were obtained from previous studies [1, 8]. The subunits of each protein were aligned using MUSCLE [26] with 100 iterations, and divergent regions were eliminated using TrimAL [27]. The alignments were concatenated by using a Perl Script (https://github.com/nylander/catfasta2phyml) for generating concatenated AcsAB, AcsABC, AcsEDC, and AcsDABCE. IQ-Tree was used for phylogenetic inference with the same parameters as above. The best models for AcsAB, AcsABC, AcsEDC, and AcsDABCE were LG+R7, LG+F+R8, LG+F+R7, and LG+F+R7, respectively. Reference hydrogenase protein sequences were selected from Greening et al. [23] and Carnevali et al. [24]. The target protein sequences were further confirmed by HydDB [28] and Pfam [29] tools. Reference sequences of ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO) large subunit were obtained from a previous study [25]. Phylogenetic trees were constructed using the methods mentioned above. All trees were visualized and annotated using iTOL [30].
Gene content comparison
Clusters of orthologous proteins were inferred by OrthoFinder 2.3.11 [31] based on a set of representative genomes from four actinobacterial classes whose members are mostly anaerobic (Ca. Geothermincolia: n = 17; Ca. Humimicrobiia: n = 7; Ca. Aquicultoria: n = 11; Coriobacteriia: n = 100). Protein families that contained only one protein from a single lineage were treated as singletons and excluded from evolutionary analysis. The Bayesian tree was constructed by MrBayes [32] with parameters (ngen=3000000 Nruns=2 Nchains=4 diagnfreq=1000 relburnin=yes burninfrac=0.25 samplefreq=100 printfreq=100). The evolutionary history of anaerobic Actinobacteria was inferred by COUNT [33] as described previously [34].
Laboratory enrichment of acetogenic bacteria
A cultivation strategy was guided by using predictive genomics [35]. Enrichment attempts of actinobacterial cells were performed under dark conditions with five treatments in the laboratory to test whether they could grow with H2 and CO2: (1) G55H: the ratio of N2:H2:CO2 was 5:4:1; (2) G55HB: the ratio of N2:H2:CO2 was 5:4:1, and the concentration of 2-bromoethanesulfonate (BES) was 20 mM; (3) G55A: the concentration of acetate was 20 mM, and the headspace gas was N2; and (4) G55C: headspace gas was N2 (control treatment); (5) G55O: stored in −80 °C (original sample). Fresh sediment slurries were passed through 2-mm sieves to homogenize and remove coarse materials. 5 mL of slurry was filled into 10 mL glass bottles with the final sediment (d.w.) to water ratio of 1:7.5. The temperature was set as 55 °C. Each treatment was carried out in triplicate. Gas samples were taken from headspace with a pressure-lock precision analytical syringe (Baton Rouge, LA, USA). The concentration of CH4 and CO2 was measured by using an Agilent 7820 A gas chromatograph, and H2 was measured by GC 6800T. Organic acid concentrations were performed by an Agilent 1200 Series liquid chromatography on day 57.
After 120 days, cells from all treatments were collected by centrifugation (10,000 × g, 20 min), and genomic DNA was extracted as described above. The Ca. Geothermincolia-specific primer set (5′-GGAAGGCCGAAGCCAACCTTT-3′, 5′-TCGTGCCGTGACGGTACCTCGG-3′) was used to determine whether the Ca. Geothermincolia existed in our enrichments. The primer set was designed by ARB software with LTPs132_SSU database [36] and verified by the RDP’s ProbeMatch [37] and NCBI. This primer set was also confirmed by sequencing purified PCR products, which showed a high similarity compared to the target 16S rRNA gene of Ca. Geothermincolia (99%). The genomic DNA was also used for 16S rRNA gene amplicon sequencing and shotgun sequencing. The V4 hypervariable region of the 16S rRNA gene was selected for generating amplicons and subsequent taxonomic analysis. Next-generation sequencing library preparations and Illumina MiSeq sequencing were conducted at GENEWIZ, Inc. (Suzhou, China). 16S rRNA gene amplicon sequence analysis was performed with QIIME [38]. The reads were joined and assigned to samples based on barcodes. Following demultiplexing and primer removal, joined reads were filtered by mean Phred quality score ≥ 20 and a minimum length of 200 bp. Operational taxonomic units (OTUs) were clustered at 97% sequence identity with VSEARCH (1.9.6) [39]. Taxonomic assignment was performed with the RDP classifier [37], using the Silva 123 database. Diversity statistics were performed using QIIME. The de novo assembly and genome binning was performed as described above. MAGs with completeness > 50% and contamination < 5% were used to confirm the organisms in the community that contain the genes for WLP. The relative abundance of each group was calculated by kaiju [40] based on reads.
Replication rate of MAGs
To investigate the replication rate of each bin in situ, iRep (index of replication) analysis was performed [41]. High-quality genomes (completeness > 75% and contamination < 2%) were compared with one another and grouped into clusters based on 98% ANI value (determined by FastANI [42]). A genome database constructed with the representative genomes (number of scaffolds < 175) of each cluster and used for iRep and functional annotation analyses. High-quality reads from each metagenome were mapped to the genome database by using Bowtie2 with default parameters [43], and iRep was calculated based on the read mapping data [41]. Function annotation analysis was performed as above. Statistical analyses and figure display were carried out in the R program (http://cran.r-project.org/).
Results and discussion
Identification of three novel classes
We first established a database of actinobacterial MAGs that contain the WLP [8] and searched for related MAGs in a hot spring metagenomic dataset and public databases. In total, we recovered 42 actinobacterial MAGs belonging to three clades, most of which (40/42) contained the WLP (fdh, fhs, folD, metF, acsA, acsB, acsC, acsD, and acsE) (Fig. 1) (Table S3); these MAGs originated from 11 hot spring sediment samples (15 MAGs) (Table 1) and publicly available metagenomes from groundwater, freshwater sediments and water, and marine environments (27 MAGs) (Fig. 1b). Phylogenetic analysis revealed that the 42 genomes, representing three distinct lineages, are well separated from the six currently defined classes of the phylum Actinobacteria [44] (Fig. 1b). These three lineages are designated as three undescribed classes by the GTDB [15]; therefore, we propose three novel classes within the phylum Actinobacteria: Ca. Geothermincolia (GTDB_id: c_RBG-13-55-18), Ca. Humimicrobiia (GTDB_id: c_UBA1414), and Ca. Aquicultoria (GTDB_id: c_UBA9087) (Fig. 1b, Tables S4 and S5, Figs. S1–S3, and Supplementary Information). To better understand their function and phylogenetic placement, we selected 35 MAGs with >75% completeness and <5% contamination for further analysis. Based on ANI values calculated by JSpecies [45], we finally identified 20 species from 35 high-quality MAGs by using 95% ANI as the cut-off [46].
Metabolic potential of the novel Actinobacteria lineages
Metabolic pathways of 35 high-quality MAGs representing these three classes were predicted. Nearly all the genomes contained complete gene sets for glycolysis and the pentose phosphate pathway (Fig. 2 and Table S3). Only one genome (Actinobacteria_bacterium GWC2_53_9) was found to contain the gene (gltA) for citrate synthase and two genomes (Actinobacteria_bacterium HGW-Actinobacteria-3 and Actinobacteria_bacterium UBA3085) contain the genes (acnA) coding for aconitate hydratase. However, none of the MAGs contain complete gene sets for tricarboxylic cycle, which renders the functioning of TCA cycle highly improbable. Furthermore, citrate lyase was not annotated in any MAG, indicating the lack of carbon fixation by the reductive TCA (rTCA) cycle. Considering that only one gene (coxB) coding for cytochrome c oxidase subunit II was present only in two species of Ca. Aquicultoria (represented by 8 MAGs), they are probably strictly anaerobic. Biosynthetic pathways for amino acids, such as glutamine, were found in all MAGs. The genes (gdhA and/or gudB) coding for glutamate dehydrogenase were only detected in the MAGs of Ca. Humimicrobiia (C2), suggesting their ability to form glutamate and NH3 from 2-oxoglutarate. All the MAGs contain the genes for homologous recombination. Genes for flagellar components were present in 19 MAGs, suggesting they are motile and/or able to attach to solid substrates where conditions are favorable. Furthermore, genes related to bacterial chemotaxis were also detected in these genomes, which ensures their ability to relocate to favorable niches by sensing environmental chemical gradients. In nitrogen metabolism, the nasA and nirB genes were detected in several MAGs of Ca. Geothermincolia, Ca. Humimicrobiia, and Ca. Aquicultoria, but other key genes, such as nasB and nirD, were absent in all MAGs. Although narGHJ genes were detected in one species of the UBA1414 group [47], no evidence showed that these genes were present in other members of the three classes. The results make dissimilatory nitrate reduction highly unlikely in these 20 species. The gene narB was only found in two species of Ca. Geothermincolia, indicating the nitrogen respirations are not characteristic of microbes of these three classes. No genetic evidence was found to suggest chemolithotrophic nitrogen oxidation, and therefore, nitrogen-based chemolithotrophy is also unlikely. The genes (dsrA and dsrB) encoding for dissimilatory sulfite reductase were detected in the MAGs of Ca. Aquicultorales (O5), suggesting that dissimilatory sulfite reduction might be performed in these MAGs. Another gene, sqr, encoding the enzyme sulfide-quinone oxidoreductase was detected in all Ca. Aquicultorales MAGs (O5), indicating possible chemolithotrophic sulfide oxidation to polysulfide. The presence of genes (pta/eutD/acyP/ackA/acdAB/acs) coding for enzymes to generate acetate from acetyl-CoA suggests they may be acetogenic (Fig. 2 and Table S3). Interestingly, the genes (K21071) for coding diphosphate-dependent phosphofructokinase were detected in all three classes (12 species), and the genes (K01622) for fructose 1,6-bisphosphate aldolase/phosphatase were detected in two classes (13 species), which indicated that these WLP-containing Actinobacteria, like other acetogens, might carry out gluconeogenesis. The genes coding for the RuBisCO-like large subunit were found in several MAGs (Table S3), and phylogenetic analysis showed that they belong to RuBisCO Form IV proteins (Fig. S4), which is consistent with a previous study [47]. The absence of a RuBisCO small subunit and phosphoribulokinase renders the functioning of the Calvin–Benson–Bassham cycle improbable. Principal coordinates analysis based on KEGG and COG annotations showed that taxonomy rather than habitat was the dominant predictor of functional potential and revealed functional differences between the three classes (Fig. S5). Notably, their common characteristics included WLP enzymes, hydrogenases, and enzymes for generating acetate, indicating that the members of these three classes are likely acetogenic bacteria that grow via the H2-dependent reduction of CO2 to acetate, a property not known to exist in Actinobacteria (Fig. 2).

Genes involved in glycolysis, pentose phosphate pathway, TCA cycle, pyruvate metabolism, WLP, sulfur metabolism, membrane transporters, and other functions are shown. The circles represent the different genes, and are colored by actinobacterial classes. Sectors colored with green, red, or yellow mean that no less than half the MAGs of Ca. Geothermincolia, Ca. Humimicrobiia, and Ca. Aquicultoria, respectively, contain this gene; the white sector means no more than one MAG contains this gene; the black sector means that no less than 2, but less than half the MAGs contain the gene. The numbers inside the circle represent the genes presented in Table S3.
When all genomes of the cultured Actinobacteria, including the 100 members of the class Coriobacteriia presented in Fig. 1b, were analyzed, all the members were found to lack the key genes for the WLP, indicating the absence of the WLP complex in the currently cultured Actinobacteria. However, analysis of the MAGs representing the three new classes revealed the widespread presence of genes encoding the WLP in the Actinobacteria. To investigate the evolutionary relationships of the CODH/ACS complex in Actinobacteria, we constructed phylogenetic trees from a series of alignments of CODH/ACS subunits (Fig. 3; Figs. S6–S8). Due to lack of representative MAGs from Actinobacteria, a previous study assigned the CODH/ACS complex of Bacteria into two large clades corresponding to “Terrabacteria” and “Gracilicutes” [1]. Our phylogeny from concatenated alignments of CODH/ACS subunits is in overall agreement with those two lineages [1]; however, the sequences from the Actinobacteria MAGs were not monophyletic with other “Terrabacteria” enzymes, indicating a more complex evolutionary history between “Terrabacteria” and “Gracilicutes” CODH/ACS genes (Fig. 3; Figs. S6–S8). The phylogeny also showed that the Actinobacteria CODH/ACS genes formed three major clades, representing the three actinobacterial classes, suggesting vertical evolution. Synteny of AcsABC subunits (Fig. S9) also suggested that the CODH/ACS genes were likely present in the common ancestor of these three actinobacterial classes and extended by vertical inheritance. When the CODH/ACS from the genomes of the uncultivated Actinobacteria were taken into consideration, a number of HGT events need to be considered to explain the topological discrepancies between the CODH/ACS trees and the phylogenomic history of the “Terrabacteria–Gracilicutes” [1, 48]. Notably, the Actinobacteria, belonging to the “Terrabacteria” might be putative donors of CODH/ACS to several members of the “Gracilicutes” [1], including members of the Nitrospirae, Deltaproteobacteria, and Thermodesulfobacteria.

The phylogenetic tree was rooted according to refs. [1, 62]. The red stars represent the classes proposed in this study. Phylogenetic groups are colored. The clades are labeled with “Terrabacteria” and “Gracilicutes” according to a previous study [1]. Notably, the concatenated AcsABC enzymes of these three actinobacterial classes (Terrabacteria) are located in the previous “Gracilicutes” clade; the deep branching position of actinobacterial enzymes suggests HGT from Actinobacteria to multiple “Gracilicutes” groups.
Insights into H2-dependent Wood–Ljungdahl carbon fixation pathway
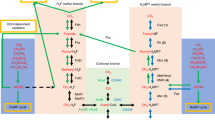
Acetogenic bacteria are both phylogenetically and physiologically diverse [6]. The pathway used for CO2 fixation (WLP) is very similar in all homoacetogens, but the electron donors and acceptors that are used for redox reactions are different. Hydrogen is a key electron donor in geothermal ecosystems [49], and is typically used for reduction of CO2 to acetate in acetogens [6]. Almost all the MAGs were predicted to encode hydrogenases, and the genes coding for hydrogenases were always close to genes for the WLP (Fig. S9), suggesting that hydrogen metabolism might be essential for a functional WLP in Actinobacteria. In total, NiFe hydrogenases (groups 1a, 1b, and 3b–3d) (Table S6), FeFe hydrogenases and energy-converting hydrogenase-related complexes (Ehr) were detected in the actinobacterial MAGs (Figs. S10–S12); yet, the complement of hydrogenases encoded by each class was distinct. The Ca. Geothermincolia, Ca. Humimicrobiia, and Ca. Aquicultoria MAGs contain genes coding for NiFe hydrogenases in group 3c, suggesting they might use this complex for hydrogen oxidation coupled with ferredoxin reduction [23]. Since only reduced ferredoxin (Fdred) can provide electrons for the reduction of CO2 to CO (the largest thermodynamic barrier in WLP [6]), and the genes coding for ferredoxin are located within the WLP cluster (Fig. S9), H2-derived Fdred is most likely the electron donor in these bacteria. In the MAGs QZM4_2.bin.157 and QZM4_3.bin.195 (Ca. Geothermincolia), the genes coding for formate dehydrogenase are preceded by genes encoding a heterodisulfide reductase complex (HdrABC-MvhD), indicating the potential for CO2 reduction to formate coupled with H2 oxidation (Fig. S9). Although the genes for MvhA could be detected in all three classes, the phylogenetic analysis showed they belong to three distinct clades (Fig. S10). In addition, class-specific hydrogenases were detected in these three lineages. For example, Ca. Aquicultoria MAGs encode NiFe group 4-related hydrogenases (Ehr) that lack typical CxxC motifs, which are suspected to couple the peripheral catalytic activity and the member charge translocation [50, 51] (Fig. S11). Notably, genes (acsA, acsB, acsC, acsD, fhs, fold, metF) coding for WLP enzymes are close to Ehr complexes, suggesting that CO2 fixation may benefit from the oxidation-reduction reactions catalyzed by this complex (Fig. S9). The possible use of Ehr complexes in WLP activity in Ca. Aquicultoria is unusual because well-studied homoacetogens use either Rnf or Ech complexes [6]. Similarly, the ATPase complexes likely to be associated with catabolic use of the WLP were distinct in the three classes (Fig. 4 and Supplementary Information). Thus, the basic process of actinobacterial homoacetogenesis from H2 and CO2 is the same, whereas the enzymes for hydrogen metabolism, energy conservation, and ATP production are different (Fig. 4). Actinobacterial acetogens might contain novel themes for energy conservation more than use the Rnf or Ech types [6], such as the “Ehr-acetogens” type. This also shows the enzyme variations of bioenergetics for WLP in acetogens.

a Cytosolic bidirectional [NiFe]-hydrogenase group 3c, Rnf complex, multicomponent Na+/H+ antiporter, two copies of V-type ATPase, and other genes (acdAB, acs, and acyP) for homoacetogenesis in Ca. Geothermincolia. b Membrane-bound H2-uptake [NiFe]-hydrogenase group 1a, cytosolic bidirectional [NiFe]-hydrogenase group 3b and 3c, Rnf complex, V-type ATPase, F-type ATPase, and other genes (pta, eutD, acyP, ackA, and acdAB) for homoacetogenesis in Ca. Humimicrobiia. c Cytosolic bidirectional [NiFe]-hydrogenase group 3c and 3d, Rnf complex or Ehr complexes, V-type ATPase, F-type ATPase, and genes (pta, acyP, ackA, acdAB, and acs) for homoacetogenesis in Ca. Aquicultoria. All the proteins presented in this figure are supported by the gene clusters within the MAGs (Fig. S9), phylogenetic analyses of hydrogenases (Fig. S10), and the functional annotation results (Table S3).
Based on the widespread absence of genes for CODH/ACS in Actinobacteria (Fig. 1b), the position and monophyly of Ca. Geothermincolia, Ca. Humimicrobiia, and Ca. Aquicultoria and the synteny of the acsABC complex, our analysis suggests that the WLP was present in the Actinobacteria before the divergence of these three classes. Considering the hydrothermal origins of the WLP [52], an ancestral member of these three actinobacterial classes may have originally obtained the WLP through an ancient lateral gene transfer event from a thermophile and expanded by vertical inheritance within actinobacterial classes and by ancient HGT events from Actinobacteria (“Terrabacteria”) to other groups (Fig. 3). Comparison of components for hydrogen metabolism shows that the three classes might utilize very different sets of hydrogenases to perform H2 oxidation. Phylogenetic analysis of hydrogenases further indicates these proteins are neither closely related among the classes nor phylogenetically congruent with those in the Coriobacteriia. The most parsimonious inference from these data is that the last common ancestor of the Actinobacteria did not encode such proteins for oxidizing H2 and that the anaerobic Actinobacteria acquired them independently after their divergence, suggesting that the common ancestor of these three classes might have been unable to perform H2-dependent WLP CO2 fixation (Fig. 5). However, with the detection of various types of hydrogenases and energy-conserving mechanisms, along with evidence that acetogens can use the WLP with a very small free energy change (less than 1 mol of ATP) [6], we hypothesize that homoacetogenesis likely occurs in Ca. Geothermincolia, Ca. Humimicrobiia, and Ca. Aquicultoria. These Actinobacteria are monophyletic and separate from the well-known aerobic actinobacterial lineages, suggesting that homoacetogenesis might be the dominant lifestyle in these three anaerobic classes.

The Bayesian tree topology was determined by MrBayes [32]. All nodes with posterior possibilities higher than 0.9 were indicated as black circles. Ancestral genome content was reconstructed from the genomes of classes Ca. Geothermincolia, Ca. Humimicrobiia, Ca. Aquicultoria, and Coriobacteriia by using COUNT software [33]. The blue star indicates the inferred root of a H2-dependent WLP CO2 fixation ancestor prior to the divergence of the three classes.
Laboratory enrichment of acetogenic Actinobacteria
Known acetogenic bacteria are able to convert substrates such as formate, pentoses, and H2/CO2 to acetate [53,54,55]. The generated acetate can be further used by methanogens and other microorganisms, which makes acetogens a vital part of the anaerobic food web [6]. In this study, our broader genomic investigation suggested homoacetogenesis in three novel actinobacterial classes. To test our hypothesis, anaerobic enrichments with different treatments were performed (Fig. 6a). In the long-term enrichment experiments conducted with 55 °C sediments from Gumingquan Hot Spring, the concentration of H2 and CO2 showed a continuous decreasing trend in treatments amended with H2 and CO2 (G55H and G55HB) (Fig. S13). This finding could be explained by the activity of H2-dependent metabolisms, such as acetogenesis and methanogenesis. After methanogenesis was suppressed by BES (G55HB), acetate accumulated compared to the treatments without BES (G55H versus G55HB) (Fig. S13). No appreciable acetate was produced in treatments without H2 (G55C) (Fig. S13). Both Ca. Geothermincolia-specific PCR and 16S rRNA gene amplicon sequencing results indicated that the target Actinobacteria could only be detected in the treatments with H2 and CO2 addition (G55H and G55HB), while they were undetectable in the original sample (G55O), control treatment (G55C), and the treatment with acetate (G55A) (Fig. 6). This suggests that the acetogenic Actinobacteria were below the detection limit in the natural sediments but that they were enriched by supplementation of H2 and CO2.

a Overview of the experimental design of the enrichment strategy. b Relative abundance of taxa in different treatments. UPGMA tree was calculated based on the Bray–Curtis distance matrix. Members containing genes coding for CODH/ACS based on MAGs from enrichment samples are shown with red stars. c Relative abundance of the key groups of acetogens or methanogens. Desulfobacterota was not detected in the 16S rRNA gene amplicon dataset.
Metagenomic analysis of these enrichment samples resulted in 101 MAGs (292 MAGs in total) coding for CODH/ACS belonging to five phyla (Methanobacteriota, Thermoproteota, Actinobacteria, Desulfobacterota, and Firmicutes) based on GTDB-Tk results (Table S7). Functional analysis based on KEGG showed that the MAGs (G55A.bin.57, G55A.bin.63, G55HB.bin.8, G55H.bin.27, G55H.bin.79, G55C.bin.56, G55C.bin.67, and G55O.bin.9) affiliated with Methanobacteriota and Thermoproteota also contain mcrABCDG, suggesting they are potential hydrogenotrophic methanogens, whereas the MAGs representing Actinobacteria (Ca. Geothermincolia), Desulfobacterota, and Firmicutes contain the genes for homoacetogenesis (Table S7). Two MAGs (G55H.bin.14 and G55HB.bin.84) belonging to Ca. Geothermincolia were obtained only from the samples treated with H2 and CO2 (G55H and G55HB) (Table S7). These two MAGs from the H2/CO2 enrichments and GMQP.bin.3 from the same environment were nearly identical (>99.7% ANI) and could be assigned to the same species (Fig. S14). This species also showed a higher abundance in G55H and G55HB (56–122 and 41–87 times higher) compared to other treatments (G55O, G55C, and G55A) based on the metagenomic dataset (Fig. S14c).
In the 16S rRNA gene amplicon datasets, Desulfobacterota OTUs were not detected in any treatment (Fig. 6b), indicating that Desulfobacterota might not provide a major contribution to homoacetogenesis. The relative abundance of Ca. Geothermincolia in the H2/CO2 treatments (G55H and G55HB) was lower than the Firmicutes (Clostridia) (Fig. 4 and Table S8). However, only Ca. Geothermincolia showed a consistent increase in the H2/CO2 treatments compared to other treatments (Fig. 6c). It is worth noting that a recent study involving long-term incubation supplemented with paraffins provided evidence for transcriptional activity of acdAB and acs genes in Actinobacteria (members of Ca. Geothermincolia) under anaerobic conditions, which supported the conversion from acetyl-CoA to acetate [9]. acdAB or acs genes are prevalent in our MAGs, indicating that these genes might be the main functional genes in producing acetate in these three classes. This confirms our predictions for Ca. Geothermincolia metabolism based on metagenomics. In addition, with the growing evidence in the G55H and G55HB treatments, these results support our hypothesis that the WLP operates reductively and that homoacetogenesis is feasible in anaerobic Actinobacteria. Comparing to other autotrophic microorganisms, metabolic flexibility is seen as a key ability of acetogenic Actinobacteria to compete in ecosystems and might explain their ubiquitous distribution.
Metabolic reconstruction in G55H and G55HB treatments
Based on the analysis of 105 representative MAGs (Table S9) obtained from metagenomic datasets from G55H and G55HB treatments, the Methanobacteria and Aquificae in the G55H cultures (average relative abundance > 1%) are potential hydrogen-/sulfur-oxidizing bacteria with capacity for carbon fixation through the WLP coupled to methanogenesis or the rTCA cycle (Fig. 7) [7, 56]. Besides Methanobacteria, there is another one MAG (G55A.bin.63), which belongs to the class Methanomethylicia, contains the complete gene cluster mcrABGCD encoding for the methyl coenzyme M reductase (Table S9), indicating its methanogenic potential. Also, members of Ca. Methanomethylicus mesodigestus, Ca. Methanomethylicus oleisabuli, and Ca. Methanosuratus petrocarbonis within this class have been proven to be hydrogenotrophic or methylotrophic methanogens [7]. The iRep value of Methanomethylicia is higher than 1.5 in G55H (Table S9), which means that, theoretically, more than half of the cells in the population are replicating [57]. The Thermoproteia G55A.bin.25 MAG is a potential sulfate-reducing archaeon that is phylogenetically closely related to the Archaeoglobi [58]. The GMQP.bin.85 belongs to Lokiarchaeia and may be a homoacetogen based on near-complete gene sets coding for CODH/ACS and acdAB, which is consistent with previous metabolic analyses of Lokiarchaeia [59,60,61]. Other potential homoacetogens were found in members of Ca. Geothermincolia, Thermodesulfobacteria, and several Firmicutes classes (Table S9). In the G55H and G55HB treatments, Ca. Geothermincolia had the highest average relative abundance (0.73 and 0.54%, respectively) compared to other homoacetogens. The iRep values (1.20 in G55H treatment and 1.21 in G55HB treatment) of Ca. Geothermincolia showed that our target microbial group kept a high replication rate. The other major microbial groups, such as Thermus, Chloroflexia, and Proteobacteria, have organotrophic potential based on genes related to carbohydrate degradation and nitrogen reduction, and no complete carbon fixation pathway could be detected in their genomes (Table S9).

The metabolic models are based on the 105 representative MAGs. Methano- Methanobacteria, M- Methanomethylicia, Loki- Lokiarchaeia, Desulf- Desulfurococcales, Thermo- Thermodesulfobacteria, Chlo- Chloroflexia, Firm- Firmicutes, P- Proteobacteria, Cy- Cyanobacteriia. The function was colored by the average iRep values that calculated by the members contain the same function in each group.
Our enrichment experiments supplemented by H2 and CO2 (G55H and G55HB) provided ideal conditions for H2-dependent chemoautotrophs. In addition, other critical factors, such as anaerobic and dark conditions, suggested the oxygen- or/and light-dependent autotrophic microorganisms may not dominate the community. Metabolic flexibility, such as possession of the WLP and capacity for utilization of organic substrates for fermentation, may allow homoacetogens to take advantage of fluctuating H2 concentrations. Thus, by utilizing the oxidation of H2 as an energy source, these putative chemoautotrophic homoacetogens very likely play essential roles in supporting thermophilic ecosystems within terrestrial hot spring environments.
Conclusions
This study provides the first insight into homoacetogenesis in three novel actinobacterial classes and suggests an important role for Actinobacteria in the evolutionary history of bacterial CODH/ACS. The results suggest that H2-dependent WLP CO2 fixation was acquired after the divergence of Ca. Geothermincolia, Ca. Humimicrobiia, and Ca. Aquicultoria through independent acquisitions of hydrogenases and energy conservation modules. The results of our first enrichment effort of Ca. Geothermincolia support the potential occurrence of homoacetogenesis in Actinobacteria and add the evidence that homoacetogenesis may be an essential metabolism in hot spring. Overall, this work expands our knowledge of the diversity and biological functions of the phylum Actinobacteria.
Data availability
The MAGs described in this paper have been deposited under NCBI PRJNA649850 and the eLibrary of Microbial Systematics and Genomics, https://www.biosino.org/elmsg/index, under accession numbers LMSG_G000001169.1–LMSG_G000001505.1. Additional raw data that support the findings of this study are available from The National Omics Data Encyclopedia (https://www.biosino.org/node) under project OEP001752 and OEP001755.
References
Adam PS, Borrel G, Gribaldo S. Evolutionary history of carbon monoxide dehydrogenase/acetyl-CoA synthase, one of the oldest enzymatic complexes. Proc Natl Acad Sci USA. 2018;115:E1166–73.
Borrel G, Adam PS, McKay LJ, Chen LX, Sierra-García IN, Sieber CM, et al. Wide diversity of methane and short-chain alkane metabolisms in uncultured archaea. Nat Microbiol. 2019;4:603–13.
Ragsdale SW, Pierce E. Acetogenesis and the Wood–Ljungdahl pathway of CO2 fixation. BBA-Proteins Proteom. 2008;1784:1873–98.
Müller B, Sun L, Schnürer A. First insights into the syntrophic acetate‐oxidizing bacteria–a genetic study. MicrobiologyOpen. 2013;2:35–53.
Schuchmann K, Müller V. Energetics and application of heterotrophy in acetogenic bacteria. Appl Environ Microbiol. 2016;82:4056–69.
Schuchmann K, Müller V. Autotrophy at the thermodynamic limit of life: a model for energy conservation in acetogenic bacteria. Nat Rev Microbiol. 2014;12:809–21.
Evans PN, Boyd JA, Leu AO, Woodcroft BJ, Parks DH, Hugenholtz P, et al. An evolving view of methane metabolism in the Archaea. Nat Rev Microbiol. 2019;17:219–32.
Vavourakis CD, Andrei AS, Mehrshad M, Ghai R, Sorokin DY, Muyzer G. A metagenomics roadmap to the uncultured genome diversity in hypersaline soda lake sediments. Microbiome. 2018;6:1–18.
Liu YF, Chen J, Liu ZL, Shou LB, Lin DD, Zhou L, et al. Anaerobic degradation of paraffins by thermophilic Actinobacteria under methanogenic conditions. Environ Sci Technol. 2020;54:10610–20.
Zhang Y, Wu G, Jiang H, Yang J, She W, Khan I, et al. Abundant and rare microbial biospheres respond differently to environmental and spatial factors in tibetan hot springs. Front Microbiol. 2018;9:2096.
Hua ZS, Han YJ, Chen LX, Liu J, Hu M, Li SJ, et al. Ecological roles of dominant and rare prokaryotes in acid mine drainage revealed by metagenomics and metatranscriptomics. ISME J. 2015;9:1280–94.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2020;36:1925–7.
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11:119.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–5.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64.
Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–8.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Kalyaanamoorthy S, Minh BQ, Wong TK, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587.
Greening C, Biswas A, Carere CR, Jackson CJ, Taylor MC, Stott MB, et al. Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J. 2016;10:761–77.
Carnevali PBM, Schulz F, Castelle CJ, Kantor RS, Shih PM, Sharon I, et al. Hydrogen-based metabolism as an ancestral trait in lineages sibling to the Cyanobacteria. Nat Commun. 2019;10:1–15.
Jaffe AL, Castelle CJ, Dupont CL, Banfield JF. Lateral gene transfer shapes the distribution of RuBisCO among candidate phyla radiation bacteria and DPANN archaea. Mol Biol Evol. 2019;36:435–46.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Søndergaard D, Pedersen CNS, Greening C. HydDB: a web tool for hydrogenase classification and analysis. Sci Rep. 2016;6:1–8.
El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427–32.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5.
Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157.
Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Csűös M. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics. 2010;26:1910–2.
Hua ZS, Wang YL, Evans PN, Qu YN, Goh KM, Rao YZ, et al. Insights into the ecological roles and evolution of methyl-coenzyme M reductase-containing hot spring Archaea. Nat Commun. 2019;10:1–11.
Jiao JY, Liu L, Hua ZS, Fang BZ, Zhou EM, Salam N, et al. Microbial dark matter coming to light: challenges and opportunities. Natl Sci Rev. 2021;8:nwaa280.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–71.
Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–42.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
Menzel P, Ng KL, Krogh A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat Commun. 2016;7:1–9.
Brown CT, Olm MR, Thomas BC, Banfield JF. Measurement of bacterial replication rates in microbial communities. Nat Biotechnol. 2016;34:1256–63.
Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:1–8.
Langmead B, Wilks C, Antonescu V, Charles R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics. 2019;35:421–32.
Salam N, Jiao JY, Zhang XT, Li WJ. Update on the classification of higher ranks in the phylum Actinobacteria. Int J Syst Evol Microbiol. 2020;70:1331–55.
Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA. 2009;106:19126–31.
Chun J, Oren A, Ventosa A, Christensen H, Arahal DR, da Costa MS, et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Micr. 2018;68:461–6.
Merino N, Kawai M, Boyd ES, Colman DR, McGlynn SE, Nealson KH, et al. Single-cell genomics of novel actinobacteria with the Wood–Ljungdahl pathway discovered in a serpentinizing system. Front Microbiol. 2020;11:1031.
Coleman GA, Davín AA, Mahendrarajah T, Spang AA, Hugenholtz P, Szöllősi GJ, et al. A rooted phylogeny resolves early bacterial evolution. 2020. https://doi.org/10.1101/2020.07.15.205187.
Spear JR, Walker JJ, McCollom TM, Pace NR. Hydrogen and bioenergetics in the Yellowstone geothermal ecosystem. Proc Natl Acad Sci USA. 2005;102:2555–60.
Vignais PM, Billoud B. Occurrence, classification, and biological function of hydrogenases: an overview. Chem Rev. 2007;107:4206–72.
Marreiros BC, Batista AP, Duarte AM, Pereira MM. A missing link between complex I and group 4 membrane-bound [NiFe] hydrogenases. BBA-Bioenerg. 2013;1827:198–209.
Martin WF. Older than genes: the acetyl CoA pathway and origins. Front Microbiol. 2020;11:817.
Daniel SL, Hsu T, Dean SI, Drake HL. Characterization of the H2-and CO-dependent chemolithotrophic potentials of the acetogens Clostridium thermoaceticum and Acetogenium kivui. J Bacteriol. 1990;172:4464–71.
Fontaine FE, Peterson WH, McCoy E, Johnson MJ, Ritter GJ. A new type of glucose fermentation by Clostridium thermoaceticum. J Bacteriol. 1942;43:701.
Drake HL, Daniel SL. Physiology of the thermophilic acetogen Moorella thermoacetica. Microbiol Res. 2004;155:869–83.
Hügler M, Hube H, Molyneaux SJ, Vetriani C, Sievert SM. Autotrophic CO2 fixation via the reductive tricarboxylic acid cycle in different lineages within the phylum Aquificae: evidence for two ways of citrate cleavage. Environ Microbiol. 2007;9:81–92.
Hou J, Sievert SM, Wang Y, Seewald JS, Natarajan VP, Xiao X, et al. Microbial succession during the transition from active to inactive stages of deep-sea hydrothermal vent sulfide chimneys. Microbiome. 2020;8:102.
Müller AL, Kjeldsen KU, Rattei T, Pester M, Loy A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi) sulfite reductases. ISME J. 2015;9:1152–65.
Spang A, Stairs CW, Dombrowski N, Eme L, Lombard J, Caceres EF, et al. Proposal of the reverse flow model for the origin of the eukaryotic cell based on comparative analyses of Asgard archaeal metabolism. Nat Microbiol. 2019;4:1138–48.
Seitz KW, Lazar CS, Hinrichs KU, Teske AP, Baker BJ. Genomic reconstruction of a novel, deeply branched sediment archaeal phylum with pathways for acetogenesis and sulfur reduction. ISME J. 2016;10:1696–705.
Orsi WD, Vuillemin A, Rodriguez P, Coskun ÖK, Gomez-Saez GV, Lavik G, et al. Metabolic activity analyses demonstrate that Lokiarchaeon exhibits homoacetogenesis in sulfidic marine sediments. Nat Microbiol. 2020;5:248–55.
Raymann K, Brochier-Armanet C, Gribaldo S. The two-domain tree of life is linked to a new root for the Archaea. Proc Natl Acad Sci USA. 2015;112:6670–5.
Acknowledgements
This study was supported by funding from the National Natural Science Foundation of China (Grant Nos 91951205, 91751206, 92051108, 32061143043, 31850410475, 31670009, and 31970122), the Natural Science Foundation of Guangdong Province, China (Grant No. 2016A030312003), Guangzhou Municipal People’s Livelihood Science and Technology Plan (Grant No. 201803030030), the U.S. National Science Foundation (DEB 1557042 and DEB 1841658), and Agricultural Science and Technology Innovation Project of the Chinese Academy of Agriculture Science (No. CAAS-ASTIP-2016-BIOMA). We thank Charlotte D. Vavourakis and Gerard Muyzer for providing the actinobacterial MAGs from metagenomes of soda lake sediments, and thank Xiao-Tong Zhang and Wen-Hui Lian for data collection. The authors would like to thank Wan Liu for the MAGs submission to the eLibrary of Microbial Systematics and Genomic (https://www.biosino.org/elmsg/index). The authors would also like to thank the Guangdong Magigene Biotechnology Co. Ltd., China, for NGS sequencing.
Author information
Authors and Affiliations
Contributions
W-JL and LC jointly conceived the study. LL, GW, E-MZ, and W-DX performed the sampling. J-YJ conceptualized the research goals under the supervision of W-JL and LC. Authors J-YJ, LL, Z-SH, and QZ performed the bioinformatics analyses. LL and J-YJ prepared the main figures. NS and AO wrote the nomenclature for the actinobacterial MAGs. LF, P-FL, and A-PL performed the enrichment experiments. J-YJ, LL, BPH, NS, AO, P-FL, B-ZF, H-CJ, RK, LC, and W-JL wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jiao, JY., Fu, L., Hua, ZS. et al. Insight into the function and evolution of the Wood–Ljungdahl pathway in Actinobacteria. ISME J 15, 3005–3018 (2021). https://doi.org/10.1038/s41396-021-00935-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-021-00935-9
This article is cited by
-
Metagenomic characterization of a novel non-ammonia-oxidizing Thaumarchaeota from hadal sediment
Microbiome (2024)
-
Roseomonas populi sp. nov., an acetate-degrading bacteria isolated from the stem of Populus tomentosa
Antonie van Leeuwenhoek (2024)
-
Culturomics- and metagenomics-based insights into the microbial community and function of rhizosphere soils in Sinai desert farming systems
Environmental Microbiome (2023)
-
Formate-induced CO tolerance and methanogenesis inhibition in fermentation of syngas and plant biomass for carboxylate production
Biotechnology for Biofuels and Bioproducts (2023)
-
A genomic catalogue of soil microbiomes boosts mining of biodiversity and genetic resources
Nature Communications (2023)
