
Abstract
Treatment of emerging RNA viruses is hampered by the high mutation and replication rates that enable these viruses to operate as a quasispecies. Declining honey bee populations have been attributed to the ectoparasitic mite Varroa destructor and its affiliation with Deformed Wing Virus (DWV). In the current study we use next-generation sequencing to investigate the DWV quasispecies in an apiary known to suffer from overwintering colony losses. We show that the DWV species complex is made up of three master variants. Our results indicate that a new DWV Type C variant is distinct from the previously described types A and B, but together they form a distinct clade compared with other members of the Iflaviridae. The molecular clock estimation predicts that Type C diverged from the other variants ∼319 years ago. The discovery of a new master variant of DWV has important implications for the positive identification of the true pathogen within global honey bee populations.
Similar content being viewed by others


Introduction
RNA viruses have rapid replication and high error rates, leading to immense diversity within each viral replication cycle (Domingo and Holland, 1997). As a result, many RNA viruses are highly genetically heterogeneous and exist within infected population structures known as quasispecies. It has been proposed that this gives these viral pathogens an increased ability to shift to a new environmental niche, such as a new host, as a suitable mutant is more likely to already exist if this opportunity arises. However, if the diversity within a quasispecies becomes too high, deleterious mutations can accumulate leading to loss of overall fitness (Clarke et al., 1993). The adaptability and host range of a virus are a function of the level of diversity found within a quasispecies (termed the quasispecies swarm size) (Schneider and Roossinck, 2001). As well as a randomly generated swarm of mutants around one variant, quasispecies can also exist as a number of master variants, each with their own swarm of random mutations (Palacios et al., 2008; Lauring and Andino, 2010). Determining the extent of genetic heterogeneity in virus populations thus has important implications for predicting and preventing emerging viral diseases.
The global decline in honey bee populations over the past few decades has been attributed to the ectoparasitic mite Varroa destructor and its affiliation with Deformed Wing Virus (DWV) type A variant (Dainat et al., 2012; Martin et al., 2012; Schroeder and Martin, 2012; Francis et al., 2013). The role of honey bees as pollinators is vital to the environment and economy, as bees are a key pollinator species for agriculture. The economic value of honey bees is estimated to be more than 225 billion US dollars worldwide (Gallai et al., 2009). DWV, a member of the single-stranded positive-sense RNA genus Iflavirus (Lanzi et al, 2006), exists as a group of closely related viruses, often considered as variants of the same species complex (Ryabov et al., 2014). The ICTV (The International Committee on Taxonomy of Viruses) database categorises DWV type A as two variants, DWV (Lanzi et al, 2006) and Kakugo virus (KV) (Fujiyuki et al., 2004). A second closely related virus Varroa destructor virus-1 (VDV-1) is also part of the genus Iflavirus, which is now designated DWV type B (Martin et al., 2012; Mordecai et al., 2015). DWV type B was designated a separate species based on a nucleotide identity to DWV type A of ∼84% that, according to demarcation criteria, is sufficient dissimilarity to warrant the creation of a new master variant (Fauquet et al., 2005). VDV-1 was originally isolated from V. destructor but has since been reported to replicate in honey bees (Ongus et al., 2004; Zioni et al., 2011) where it has been shown to cause wing deformities in bees (Zioni et al., 2011). DWV type B was also recently found to dominate the DWV population in honey bees from an isolated apiary in Swindon, UK (Mordecai et al., 2015); to the apparent exclusion of any other master variants. In honey bee populations that have never been exposed to Varroa mites, DWV exists in a very large variant swarm with numerous master variants (Martin et al., 2012). However, transmission of DWV by Varroa reduces variant diversity to one master variant (Martin et al., 2012). Therefore, DWV exists as an endlessly mutating swarm of variants with these master variants constituting part of this quasispecies and sharing a recent common ancestor (Baker and Schroeder, 2008; Martin et al., 2012). The dominance of one master variant over another will lead to ultimately different life histories for the colony, that is, death if DWV type A dominates (Martin et al., 2012) or health if DWV type B dominates (Mordecai et al., 2015).
By existing as a diverse swarm of variants, viruses are able to co-occupy several biological niches. Certain biological traits may allow a virus to infect one cell type over another, known as cell tropism (Koyanagi et al., 1987). Whether or not a virus is able to infect a susceptible cell depends firstly on recognition of a cellular receptor on the cell surface and secondly on intracellular host factors that dictate whether the host cell is permissive to virus replication. Therefore, amino acid substitutions caused by nucleotide mutations in the structural or nonstructural region of the virus genome can affect both the host range and cell tropism of a virus. When categorising viruses based on a phylogenetic relationship it is important to note that a single amino acid change can have a substantial effect on the phenotypic traits of a virus. Therefore, when a virus exists as a collection of variants or quasispecies, although the phylogeny and ancestry of the viruses may be similar, the host range, tropism, pathogenicity and epidemiology of the variants may differ greatly (Domingo et al., 2012). In addition, recombination between these variants is a source of further variation (Moore et al., 2011).
There are numerous biological implications of quasispecies occupying large amounts of sequence space that challenge the treatment and control of both established and emerging infectious diseases (Gomez et al., 1999). For example, quasispecies theory has been used to describe how viruses such as Hepatitis C virus and HIV are able to escape host immune responses (Pavio and Lai, 2003; Woo and Reifman, 2012). In addition, effective vaccines are yet to be developed for both these viruses because of the rapid emergence of resistant mutants under vaccine selection pressure (Gaschen et al., 2002; Law et al, 2013). For similar reasons, the highly divergent nature of RNA viruses has been implicated in the emergence of antiviral drug-resistant infections in AIDS (Metzner et al., 2009), hepatitis C (Halfon and Locarnini, 2011), hepatitis B (Nishijima et al., 2012) and influenza (Boivin et al., 2002).
Conventional methods to define and analyse variance within RNA viruses include RNA ‘fingerprinting’ (Domingo et al., 1978) and reverse transcriptase-PCR amplification using specifically designed primers (see, for example, Highfield et al., 2009). Clone libraries and Sanger sequencing were used to identify the DWV type A as being associated with Varroa infestation and colony collapse (Martin et al., 2012). Although these techniques are valid for identifying known variants, primer-based methods are prone to missing unidentified variants and are biased towards overrepresented sequences (Gomez et al., 1999). PCR-based methods are less appropriate to determine the extent of variation in a quasispecies that is not normally distributed that is, where multiple variants exist, each with their own spectrum of mutants (Gomez et al., 1999). In these instances deep sequencing methods such as Illumina platforms are more suited to discovering new variants as well as to diversity analysis (Wood et al., 2014).
Illumina sequencing is advantageous for samples with large amounts of genetic variation because of its depth of sequencing, although significant raw read analysis is required. Reference-based assembly methods can overlook biological variants because of inaccurate read alignments and loss of data (Archer et al., 2010; Iqbal et al., 2012; Yang et al., 2012), although former studies using high-throughput next-generation sequencing of DWV have used these methods (Moore et al., 2011; Ryabov et al., 2014).
A previous study (Highfield et al., 2009) found that despite controlling Varroa populations, high DWV loads were associated with overwintering colony losses (OCL). Historical losses due to OCL of ∼10% were normal; these have now risen to ∼20% since the establishment of Varroa. This suggests that a ‘new’ non-Varroa-transmitted DWV master variant may be circulating in some colonies during the winter causing OCL. To investigate this hypothesis we used a bespoke de novo assembly pipeline (Mordecai et al., 2015). The Vicuna (broadinstitute.org/scientific-community/science/projects/viral-genomics/vicuna) de novo assembler was used as it is designed to assemble highly heterogeneous viral populations and is well suited to the computational challenge that the DWV quasispecies present (Yang et al., 2012). As well as assembling the DWV type A master variant, a third DWV master variant (Type C) was assembled and confirmed to be distinct through phylogenetic inference.
Materials and methods
Sequencing and assembly
Total RNA was sequenced without an amplification step. Illumina (San Diego, CA, USA) Hi-seq (2 × 100) pair-end sequencing was carried out by TGAC (The Genome Analysis Centre) and at the University of Exeter on four samples from Highfield et al. (2009); Late June, Early October and Late October from GD1, and early October from GD2 (Supplementary Table S1). The samples were originally collected from Devon in the southwest of England (Highfield et al., 2009). Twenty asymptomatic bees were pooled for each sample before RNA extraction. RNA extractions were prepared as in Highfield et al. (2009) followed by a complementary DNA amplification step before sequencing.
A Bioinformatics pipeline was developed to accommodate the large amount of variation found within the DWV species complex. First, the quality of the raw reads was verified using FastQC (Babraham Bioinformatics, Cambridge, UK). Samples were then converted from fastq to fasta using the fastq_to_fasta script that is part of the FASTX-toolkit (Hannon Lab) (http://hannonlab.cshl.edu/fastx_toolkit/).
To isolate the DWV complex sequence reads from the host and other contaminating sequences, the BLASTn (Altschul et al., 1990) tool was used. The reads were searched against a custom BLAST database containing the DWV, VDV-1 and KV genomes, with an e-value of 10e−5. BLAST was carried out against Read 1 of the Illumina data. The ncbi-blast-parser perl script (http://www.bioinformatics-made-simple.com/2012/07/ncbi-blast-parser-extract-query-and.html) was then used to parse and read the top hit of the BLAST output.
Next, ‘sed’ and ‘awk’ scripts were used to delete empty lines and the reads that contained ‘nohits’. The corresponding BLAST hits were extracted from the Read 2 raw reads using QIIME (Caporaso et al., 2010). The paired reads were balanced using a custom script written in R version 3.2.0 (R Core Team, 2015). Finally, the balanced DWV family reads were assembled using the Vicuna assembler that was developed to generate consensus assemblies from genetically heterogeneous populations, specifically RNA viruses. (Yang et al., 2012).
Vicuna contigs >200 bp in length were imported into Geneious (Version 7.04, created by Biomatters, Auckland, New Zealand) and the ‘Map to Reference tool’ was used to align the contigs with the DWV and VDV-1 reference genomes. For several of the samples the Vicuna assembly yielded full-length contigs that covered the whole genome, whereas for others consensus scaffolds were created from two or more contigs. The ends of the contigs were then trimmed of any assembly or sequencing artefacts.
A second novel variant was also assembled by Vicuna. In order to create a consensus sequence of the novel variant, the contigs from three samples were realigned against the DWV genome. Any contigs containing sequence other than the novel variant were removed. If a contig contained the novel variant as well as sequence belonging to the type A DWV genome because of recombination or in silico recombination, the type A DWV regions were trimmed and deleted. A consensus genome of the novel variant was created from these sorted and trimmed contigs. Accordingly, the same steps were carried out on the type A sequence in order to assemble the type A genome consensus from Devon. The DWV scaffolds were then aligned with full-length genome sequences from the NCBI (National Center for Biotechnology Information) database using the MUSCLE alignment tool (Edgar, 2004) within Geneious and the full-length genome was obtained. Full genome comparisons were visualised in mVISTA (Frazer et al., 2004).
To quantify the number of reads attributed to each DWV variant using BLAST (Supplementary Table S1), the novel Type C variant genome sequence was added to the custom DWV family database and the BLAST for each sample was carried out a second time. This allowed the Type C variants to be categorised correctly by BLAST rather than attributed to the closets hit to the three other reference genomes.
To validate the quantification of reads attributed to each variant, the balanced read 1 and 2 BLAST hits were aligned competitively to three reference genomes using the ‘Map to Reference Tool’ within Geneious.
Consensus genomes of type A and Type C were translated using the ‘live annotation’ tool within Geneious. Because of repetitive regions in the genome, assembly error took place in several sites, leading to an incomplete open reading frame. These assembly errors were corrected by using the Vicuna analysis tool to pull out individual reads covering these regions, and the correct sequence was determined. Finally, the RDP4 programme (Martin et al., 2010) was used to determine whether any recombination took place between the sequences. Assembled genomes are available from the European Nucleotide Archive under the accession numbers ERS657948 (type A) and ERS657949 (Type C).
Amino acid-based phylogeny
To establish the phylogenetic relationship of DWV subtypes and closely related Iflaviruses, we reconstructed the phylogeny of the conserved RdRp amino acid sequences for seven DWV subtypes, spanning all three types that were either sequenced and assembled from the Devon hive or available from genbank (type A: NC_005876.1, NC_004830.2; type B: KC_786222.1, NC_006494.1, JQ_413340), as well as Formica exsecta Virus 1 (NC_023022.1) and Sacbrood Virus (NC_002066.1). We used a Bayesian approach using MrBayes (v. 3.1.2) (Huelsenbeck and Ronquist, 2001). We assumed a fixed rate model of protein evolution and reconstructed the phylogeny using a model jumping method. This method allows for different models of amino acid substitution to be used in the Markov chain Monte Carlo (MCMC) procedure, with all models contributing to the final result weighted according to their respective posterior probability. We ran two runs of four chains for 4 000 000 MCMC generations, sampling trees every 1000 generations. All trees were drawn using FigTree v.1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
Evolutionary rate
To estimate the evolutionary rate of DWV and its subtypes, we collated three independent data sets with temporal information for at least one population: (A) partial lp-gene; 10 populations, n=78, 328 nt (1323–1650 bp) (Genbank AJ489744, AY292384, GU109335, HM162355, JF346615-JF346620, JF346624-JF346629, JF346633-JF346639, KF164292, KF164293, KJ437447, KP734726, KP734738, KP734747, KP734765-KP734770, KP734774-KP734787, KP734817-KP734825, KP734827-KP734846); (B) partial capsid-gene; 1 population, n=167, 1215 nt (2634–3848 bp) (Genbank AY292384, HQ655502-HQ655561, KF314827- KF314932); (C) partial RdRp-gene; 1 B. terrestris population, n=145, 508 nt (8016–8522 bp) (Genbank KP734326–KP734470). To test whether these fragments contain a molecular clock signal, we estimated the root-to-tip divergence using Path-o-gen v.1.4 (http://tree.bio.ed.ac.uk/software/pathogen/) to estimate how much genetic variation can be explained by the sampling date. We constructed maximum likelihood phylogenetic trees without the assumption of a molecular clock in Phylip v. 3.695 (evolution.genetics.washington.edu; Felsenstein, 1989) and tested whether the regression between root-to-tip distance in these maximum likelihood trees and the age of the samples indicated a clock-like signal. In addition, we tested for a significant temporal signal by randomising the temporal information across each data set 100 times and compared the resulting random evolutionary rates from BEAST with the real data set. A temporal signal is supported if there is a significant difference between the real data set and the randomised data sets (Ramsden et al., 2009; Alizon and Fraser, 2013). Both analyses support a temporal signal in these data sets (Supplementary Figure S3 and Supplementary Table S4).
Model selection and BEAST runs
To determine the appropriate molecular clock models for each data set, we used the path sampling maximum likelihood estimator implemented in BEAST 1.8 (Baele et al., 2012, 2013). As this method is very computationally intensive, we first used a range of simpler tests to limit the number of models to be compared by this method as suggested by Alizon and Fraser (2013), and Drummond and Bouckaert (2014). For the three large temporal data sets, we used jModelTest v.2.1.1 (https://github.com/ddarriba/jmodeltest2; Posada, 2008) to compare substitution models based on the Bayesian Information Criterion (Alizon and Fraser 2013); the resulting substitution models were the Tamura-Nei (TnR) model (lp-fragment) and the Hasegawa, Kashino and Yano (HKY) model (vp3- and RdRp-fragments), both with Gamma variation. For the DWV-subtype data set, we compared the general time-reversible model and the SRD06 model (Shapiro et al., 2006) using path sampling. We partitioned substitution rates between the first and second and third codon positions as, for all data sets, the third codon position had a significantly higher rate. We tested whether a strict clock rate can be excluded by running models with a lognormal relaxed clock; if the relaxed clock’s coefficient of variation statistic abuts the zero boundary, a strict clock cannot be excluded (Gray et al., 2011). We ran models with exponential population growth for all models; if the exponential growth rate was significantly higher than zero, a constant population size can be excluded. We then used path sampling to distinguish between clock models (exponential and lognormal relaxed clock) and demographic models (constant population size, exponential population growth or a Gaussian Markov random field (GMRF skyride); Drummond et al., 2002; Minin et al., 2008) as indicated for each fragment by the initial analyses. Based on these analyses, we chose an exponential relaxed clock and exponential growth prior for the lp- and capsid-fragments and a lognormal relaxed clock and constant growth prior for the RdRp-fragment as well as for the DWV-subtype analyses.
To generate a genome-wide estimate for the evolutionary rate in DWV, we calculated the mean of the relaxed clock means for the individual fragments (lp-fragment: 9.097 × 10−4 (95% highest posterior density: 4.412 × 10−4–1.394 × 10−3), capsid-fragment: 1.845 × 10−3 (1.159 × 10−3–2.569 × 10−3), RdRp-fragment: 1.278 × 10−3 (4.131 × 10−4–2.513 × 10−3), resulting in a mean evolutionary rate of 1.346 × 10−3 (5.41 × 10−4–2.627 × 10−3) substitutions/site/year. We implemented this evolutionary rate as an uncorrelated lognormal clock prior for the DWV-subtype analysis with a lognormal distribution with a mean of 1.35 × 10−3 in real space and a log s.d. of 0.4. We used the default priors in BEAST v.1.8.1. We ran models with 2 runs each of 50 million MCMC generations, sampling every 5000 generations with a burn-in of 5 million generations to obtain effective sample sizes >200. We examined traces for convergence using Tracer v.1.6 (http://www.tree.bio.ed.ac.uk/software/tracer/) and used TreeAnnotator v.1.8 (beast.bio.ed.ac.uk/downloads) to produce a Maximum Clade Credibility tree for the DWV-subtype analysis. We used the method from Xia et al. (2003), as implemented in DAMBE, to confirm that the alignment had not reached substitution saturation.
Results and discussion
Illumina Hi-seq (2 × 100) pair-end sequencing of asymptomatic honey bees from Devon was carried out on colonies that either survived (GD2) or collapsed (GD1) because of OCL (Highfield et al., 2009). To assess DWV diversity within the samples the raw reads were searched against a custom DWV family database (including KV and VDV-1 genomes) and reads matching to any of the DWV genomes were extracted. The coverage for the number of reads that matched to a custom DWV BLAST database was estimated using the Lander/Waterman equation (that is, the depth of sequencing). Genome coverage depth ranged from 457 to 165 927 × (average coverage was 86 838 ×) (Supplementary Table S1).
De novo assembly of the Illumina reads yielded a complete genome of a type A variant as well as of a novel DWV variant that we named Type C (Supplementary Table S2). Competitive alignment to the now three master variants revealed that out of the ~30 million DWV blast positive reads, ~27 million were assembled to one of three genomes (Supplementary Table S3 and Supplementary Figure S1). Around 3 million assembled to type A, ~3.5 thousand to type B and ~24 million to Type C. As expected, more reads align to the 3′ region of the genome than the 5′; an artefact of the reverse transcription 3′ bias (Brooks et al., 1995). Although reads aligned to the whole of the Type C genome, the depth of coverage was lower at the 5′ region (Supplementary Figure S1). In addition, coverage of the 5′ region of the type A variant was unusually high, indicating that this disproportionate coverage in the 5′ regions of type A and C could be the evidence of recombination between the two variants. Because of the low read depth and coverage for type B in our samples, a full genome could not be assembled (Supplementary Table S3 and Supplementary Figure S1). Nonetheless, closer examination of 3′ region where the RNA-dependent RNA polymerase (RdRp) gene is located (Baker and Schroeder, 2008) revealed that the Devon type B variant shared 100% identity to VDV-1 type B genome in this region (Supplementary Figure S2). The sequence identity of our newly assembled type A and C variants was compared with other members of the DWV complex (Table 1 and Figure 1). Both type B and C differ from the type A nucleotide sequence in similar regions of the genome. However, type B and C share only 79% nucleotide identity in the polyprotein encoding region of the genome and 89% identity in the amino acid sequence.

Plot showing the percentage identity across the whole genome of (1) Devon DWV type A (ERS657948), (2) KV type A (NC0058762), (3) VDV-1 type B (AY251269) and (4) Devon Type C (ERS657949) compared with the DWV type A (NC004830) reference genome. Plot created by mVISTA (Frazer et al., 2004).
Phylogenetic analysis showed the relationship of the novel type A and C variants from Devon to previously sequenced viruses (Figures 2 and 3). The type A genome clustered with other type A variants, whereas Type C formed a distinct and separate branch, thereby confirming the originality and thus new master variant assignment of DWV Type C. Given that type A and C can recombine with each other, we screened for any genomes with evidence of recombination and excluded them from the analysis in order to comply with the assumptions underlying phylogenetic reconstruction.

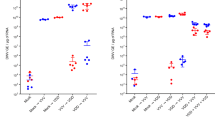
Bayesian inference of phylogeny based on a conserved region of amino acid sequence encoding for the RNA-dependent RNA polymerase, with the Bayesian support values shown on the nodes. Samples from this study are labelled with an asterisk (*). Bar represent number of nucleotide substitutions per site.

Bayesian phylogeny of the polyprotein encoding nucleotide sequence using an MCMC model (BEAST). Node labels show the age and the posterior probability in brackets. The branches are labelled with the clock rate as a relaxed clock rate was used. Genomes sequenced from this study are labelled with an asterisk (*) and are available from the European Nucleotide Archive under the accession numbers ERS657948 (type A) and ERS657949 (Type C). Scale bar represents years based on a relaxed molecular clock.
The Type C genome codes for a full-length polyprotein and translation of the genome permitted the amino acid sequence to be compared with more distant members of the genus Iflavirus (Figure 2). The relationship of the DWV complex with F. exsecta virus 2 and Sacbrood virus was analysed using a Bayesian inference of phylogeny of a conserved region of the RdRp amino acid sequence (Figure 2). The DWV sequences were attributed to three main groups (type A, B and C). The nucleotide consensus sequences created by Vicuna for type A was found to be 98.2% identical to the type A reference DWV genome. The analysis suggested that the new Type C genome is clearly related to type A and B, and forms a distinct clade to other members of the family Iflaviridae (for example, Sacbrood virus) that are all only distantly related to a dicistrovirus outgroup, F. exsecta virus.
In addition, a Bayesian analysis of the polyprotein encoding region of the sequences was carried out using an MCMC model, permitting a molecular clock model to be run within BEAST v1.8.1 (Figure 3) (Drummond et al., 2012). Divergence times were calculated based on a tip-dated coalescent model, with an evolutionary rate prior based on three independent tip-dated fragments of DWV type A. The samples in the tree span 11 years (2000–2011), and the samples used for estimating the evolutionary rate span 11, 13 and 22 years for the RdRp-, capsid- and lp- fragments, respectively. The Bayesian tree of the nucleotide sequences had a similar structure to the amino acid tree, showing that the Type C viral variant is distinct from type A and B. The molecular clock estimation predicts that Type C diverged from the other DWV variants 319 years ago (57–1010 95% highest posterior density), and type A and B disassociated from each other 181 years ago (38–497 95% highest posterior density). This estimate is unlikely to be biased by substitution saturation, as there is no evidence for saturation in our data set (Xia et al., 2003).
The phylogenetic analysis concludes that the DWV quasispecies is made up of three distinct master variants. However, the number of variants is not exhaustive and it is conceivable that more variants will be added in the future as the use of NGS becomes increasingly prolific. The result further suggests that Type C has not recently emerged, but rather is an established DWV variant. Moreover, using reverse transcriptase-PCR to amplify a region of the DWV RdRp gene, Martin et al. (2012) attributed a novel variant to the type B swarm of variants, although it is now clear that this novel variant was in fact Type C. The presence of Type C in Hawaii implies that this variant is widespread and not specific to the United Kingdom. It also confirms that it has not emerged recently as the Hawaiian sample originates from Kauai that has yet to be colonised by Varroa (Martin et al., 2012), suggesting Type C is part of the wild-type DWV quasispecies.
The high levels of heterogeneity within viral RNA populations mean that viruses are able to occupy large areas of sequence space and consequently are able to exist in multiple hosts (Domingo and Holland, 1997). DWV is a ‘generalist’ known to infect bumblebees (Genersch et al, 2006; Fürst et al., 2014), V. destructor (Ongus et al., 2004) and other insects (reviewed in Manley et al., 2015). Further work is required to ascertain whether Type C causes acute wing deformities in honey bees or its presence is because of viral ‘spill over’ from another host. Three of the four samples in this study were taken from a hive (GD1) that went on to collapse from OCL. However, as type A and the type A–C 5′ recombinant were also present, it remains unclear which variant was responsible for OCL. It is worth noting however that between 85% and 98% of the reads in hive GD1 were Type C that make up the Type C or the A–C recombinant genomes (Supplementary Table S1). The second hive (GD2) that survived OCL contained roughly equal levels of type A and C reads (which includes reads that make up the A–C recombinant); yet, the coverage indicates that the viral load was much lower in this hive. Moreover, the low abundance of DWV type B in these Devon colonies appears to confirm the observation made in the colonies from Swindon (Mordecai et al., 2015); that is, DWV type B is not present at sufficient levels to protect the honey bee from the virulent type A, or possibly C variants (as well as any recombinants between A and C). In addition, Varroa mites in the Devon colonies were controlled using chemical methods, thereby potentially preventing the mites from transmitting type B into the honey bees; as observed in the Swindon Apiary (Mordecai et al., 2015).
A recombinant between DWV type A and B has been reported previously (Moore et al., 2011), and found to be hypervirulent as it was more efficient at replicating than other variants when co-injected directly into the haemolymph of honey bee larvae (Ryabov et al., 2014). As it is presently unclear whether the Type C variant or the A–C recombinant lead to disease, further direct manipulation experiments such as those carried out by Ryabov et al. (2014 could be crucial in determining whether the novel Type C genome and any of its recombinants are hypervirulent.
The large number of DWV Type C and A–C recombinant reads in the Devon samples (Supplementary Table S1 and Supplementary Figure S1) suggests that these variants are able to replicate in honey bees. The capacity of DWV to exist as a swarm of recombining variants and occupy large amounts of sequence space may be one of the factors contributing to its capability to maintain a persistent infection. Defining DWV as a quasispecies with discrete master variants is comparable with the current categorisation of several other RNA viruses. The genetic variability within Hepatitis C virus has been classified into four hierarchical strata: genotypes, sub-genotypes, isolates and finally background variation of the quasispecies (Farci and Purcell, 2000). A total of seven Hepatitis C virus genotypes exist to date, differing from each other by >15% over their complete coding region (Smith et al., 2014). Similarly, Japanese encephalitis virus is currently classified into five genotypes based on sequence identity that differ in replication efficiency in a range of hosts (Han et al., 2014). In order to further our understanding of DWV and the impact of the virus on honey bees as well as other hosts, a more progressive classification of the DWV quasispecies would be prudent, that is, the new classification of DWV as a quasispecies with at least three master variants that can recombine with each other.
Bioinformatics pipelines designed for eukaryotes and prokaryotes are not necessarily suited to studying viral systems. For example, pipelines incorporating reference assemblers could hinder the discovery of novel virus variants. The genome sequences presented here are representative of the different variants found within a sample; however, the Vicuna pipeline is not suited to analysing the further level of diversity around these variants, for which diversity models are better suited, such as those developed by Wood et al. (2014).
As well as being of significance to globally important honey bee health, the ability to study highly heterogeneous virus genomes is of wider importance. Cross-species virus transmission and emergence of new epidemic diseases such as severe acute respiratory syndrome, Ebola and influenza are major threats to public health (Parrish et al., 2008). Exploring the extent of viral diversity in RNA quasispecies, of which DWV may be a suitable model, may offer insight into the mechanisms by which viruses are able to transmit between different hosts as well as how viruses are able to develop resistance to antiviral therapies (Domingo et al., 2012). Further study of the DWV quasispecies may help to explain how DWV is able to exist as multiple variants in many hosts, and may elucidate mechanisms by which it establishes a persistent infection among several hosts but only proves pathogenic in some.
References
Alizon S, Fraser C . (2013). Within-host and between-host evolutionary rates across the HIV-1 genome. Retrovirology 10: 49.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ . (1990). Basic local alignment search tool. J Mol Biol 215: 403–410.
Archer J, Rambaut A, Taillon BE, Harrigan PR, Lewis M, Robertson DL . (2010). The evolutionary analysis of emerging low frequency HIV-1 CXCR4 using variants through time—an ultra-deep approach. PLoS Comput Biol 6: e1001022.
Baele G, Lemey P, Bedford T, Rambaut A, Suchard MA, Alekseyenko AV . (2012). Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol Biol Evol 29: 2157–2167.
Baele G, Li WLS, Drummond AJ, Suchard MA, Lemey P . (2013). Accurate model selection of relaxed molecular clocks in Bayesian phylogenetics. Mol Biol Evol 30: 239–243.
Baker AC, Schroeder DC . (2008). The use of RNA-dependent RNA polymerase for the taxonomic assignment of picorna-like viruses (order Picornavirales) infecting Apis mellifera L. populations. Virol J 5: 10.
Boivin G, Goyette N, Bernatchez H . (2002). Prolonged excretion of amantadine-resistant influenza A virus quasi species after cessation of antiviral therapy in an immunocompromised patient. Clin Infect Dis 34: e23–e25.
Brooks EM, Sheflin LG, Spaulding SW . (1995). Secondary structure in the 3′ UTR of EGF and the choice of reverse transcriptases affect the detection of message diversity by RT-PCR. Biotechniques 19: 806–812 814–815.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Clarke DK, Duarte EA, Moya A, Elena SF, Domingo E, Holland J . (1993). Genetic bottlenecks and population passages cause profound fitness differences in RNA viruses. J Virol 67: 222–228.
Dainat B, Evans JD, Chen YP, Gauthier L, Neumann P . (2012). Dead or alive: deformed Wing Virus and Varroa destructor reduce the life span of winter honeybees. Appl Environ Microbiol 78: 981–987.
Domingo E, Holland JJ . (1997). RNA virus mutations and fitness for survival. Annu Rev Microbiol 51: 151–178.
Domingo E, Sabo D, Taniguchi T, Weissmann C . (1978). Nucleotide sequence heterogeneity of an RNA phage population. Cell 13: 735–744.
Domingo E, Sheldon J, Perales C . (2012). Viral quasispecies evolution. Microbiol Mol Biol Rev 76: 159–216.
Drummond AJ, Bouckaert R . (2014). Bayesian Evolutionary Analysis with BEAST 2. Cambridge University Press: Cambrige, UK.
Drummond AJ, Suchard MA, Xie D, Rambaut A . (2012). Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29: 1969–1973.
Drummond AJ, Nicholls GK, Rodrigo AG, Solomon W . (2002). Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161: 1307–1320.
Edgar RC . (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32: 1792–1797.
Farci P, Purcell RH . (2000). Clinical significance of hepatitis C virus genotypes and quasispecies. Semin Liver Dis 20: 103–126.
Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA . (eds). (2005) Virus Taxonomy Academic Press: San Diego, pp 739–1128.
Felsenstein J . (1989). Phylogeny Inference Package (Version 3.2). Cladistics 5: 163–166.
Francis RM, Nielsen SL, Kryger P . (2013). Varroa-virus interaction in collapsing honey bee colonies. PLoS One 8: e57540.
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I . (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res 32: W273–W279.
Fujiyuki T, Takeuchi H, Ono M, Ohka S, Sasaki T, Nomoto A et al. (2004). Novel insect picorna-like virus identified in the brains of aggressive worker honeybees. J Virol 78: 1093–1100.
Fürst MA, McMahon DP, Osborne JL, Paxton RJ, Brown MJF . (2014). Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature 506: 364–366.
Gallai N, Salles J-M, Settele J, Vaissière BE . (2009). Economic valuation of the vulnerability of world agriculture confronted with pollinator decline. Ecol Econ 68: 810–821.
Gaschen B, Taylor J, Yusim K, Foley B, Gao F, Lang D et al. (2002). Diversity considerations in HIV-1 vaccine selection. Science 296: 2354–2360.
Genersch E, Yue C, Fries I, de Miranda JR . (2006). Detection of Deformed wing virus, a honey bee viral pathogen, in bumble bees (Bombus terrestris and Bombus pascuorum) with wing deformities. J Invertebr Pathol 91: 61–63.
Gomez J, Martell M, Quer J, Cabot B, Esteban JI . (1999). Hepatitis C viral quasispecies. J Viral Hepatitis 6: 3–16.
Gray RR, Parker J, Lemey P, Salemi M, Katzourakis A, Pybus OG . (2011). The mode and tempo of hepatitis C virus evolution within and among hosts. BMC Evol Biol 11: 131.
Halfon P, Locarnini S . (2011). Hepatitis C virus resistance to protease inhibitors. J Hepatol 55: 192–206.
Han YW, Choi JY, Uyangaa E, Kim SB, Kim JH, Kim BS et al. (2014). Distinct dictation of Japanese encephalitis virus-induced neuroinflammation and lethality via triggering TLR3 and TLR4 signal pathways. PLoS Pathog 10: e1004319.
Highfield AC, El Nagar A, Mackinder LCM, LM-LJ Noel, Hall MJ, Martin SJ et al. (2009). Deformed Wing Virus implicated in overwintering honeybee colony losses. Appl Environ Microbiol 75: 7212–7220.
Huelsenbeck JP, Ronquist F . (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755.
Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G . (2012). De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat Genet 44: 226–232.
Koyanagi Y, Miles S, Mitsuyasu R, Merrill J, Vinters H, Chen I . (1987). Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science 236: 819–822.
Lanzi G, de Miranda JR, Boniotti MB, Cameron CE, Lavazza A, Capucci L et al. (2006). Molecular and biological characterization of Deformed Wing Virus of honeybees (Apis mellifera L.). J Virol 80: 4998–5009.
Lauring AS, Andino R . (2010). Quasispecies theory and the behavior of RNA viruses. PLoS Pathog 6: e1001005.
Law LMJ, Landi A, Magee WC, Lorne Tyrrell D, Houghton M . (2013). Progress towards a hepatitis C virus vaccine. Emerg Microbes Infect 2: e79.
Manley R, Boots M, Wilfert L . (2015). Emerging viral disease risk to pollinating insects: ecological, evolutionary and anthropogenic factors. J Appl Ecol 52: 331–340.
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P . (2010). RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26: 2462–2463.
Martin SJ, Highfield AC, Brettell L, Villalobos EM, Budge GE, Powell M et al. (2012). Global honey bee viral landscape altered by a parasitic mite. Science 336: 1304–1306.
Metzner KJ, Giulieri SG, Knoepfel SA, Rauch P, Burgisser P, Yerly S et al. (2009). Minority quasispecies of drug-resistant HIV-1 that lead to early therapy failure in treatment-naive and -adherent patients. Clin Infect Dis 48: 239–247.
Minin VN, Bloomquist EW, Suchard MA . (2008). Smooth skyride through a rough skyline: Bayesian coalescent-based inference of population dynamics. Mol Biol Evol 25: 1459–1471.
Moore J, Jironkin A, Chandler D, Burroughs N, Evans DJ, Ryabov EV . (2011). Recombinants between Deformed wing virus and Varroa destructor virus-1 may prevail in Varroa destructor-infested honeybee colonies. J Gen Virol 92: 156–161.
Mordecai GJ, Brettell L, Martin SJ, Dixon D, Jones IM, Schroeder DC . (2015). Superinfection exclusion in honey bees explains long-term survival of Varroa infested colonies. ISME J; e-pub ahead of print 27 October 2015; doi:10.1038/ismej.2015.186.
Nishijima N, Marusawa H, Ueda Y, Takahashi K, Nasu A, Osaki Y et al. (2012). Dynamics of Hepatitis B Virus quasispecies in association with nucleos(t)ide analogue treatment determined by ultra-deep sequencing. PLoS One 7: e35052.
Ongus JR, Peters D, Bonmatin J-M, Bengsch E, Vlak JM, van Oers MM . (2004). Complete sequence of a picorna-like virus of the genus Iflavirus replicating in the mite Varroa destructor. J Gen Virol 85: 3747–3755.
Palacios G, Hui J, Quan PL, Kalkstein A, Honkavuori KS, Bussetti AV et al. (2008). Genetic analysis of Israel Acute Paralysis Virus: distinct clusters are circulating in the United States. J Virol 82: 6209–6217.
Parrish CR, Holmes EC, Morens DM, Park E-C, Burke DS, Calisher CH et al. (2008). Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol Mol Biol Rev 72: 457–470.
Pavio N, Lai MMC . (2003). The hepatitis C virus persistence: how to evade the immune system? J Biosci 28: 287–304.
Posada D . (2008). jModelTest: phylogenetic model averaging. Mol Biol Evol 25: 1253–1256.
R Core Team. (2015) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria.
Ramsden C, Holmes EC, Charleston MA . (2009). Hantavirus evolution in relation to its Rodent and insectivore hosts: no evidence for codivergence. Mol Biol Evol 26: 143–153.
Ryabov EV, Wood GR, Fannon JM, Moore JD, Bull JC, Chandler D et al. (2014). A virulent strain of Deformed Wing Virus (DWV) of honeybees (Apis mellifera prevails after Varroa destructor-mediated, or in vitro, transmission. PLoS Pathog 10: e1004230.
Schneider WL, Roossinck MJ . (2001). Genetic diversity in RNA virus quasispecies is controlled by host-virus interactions. J Virol 75: 6566–6571.
Schroeder DC, Martin SJ . (2012). Deformed wing virus: the main suspect in unexplained honeybee deaths worldwide. Virulence 3: 589–591.
Shapiro B, Rambaut A, Drummond AJ . (2006). Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol Biol Evol 23: 7–9.
Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT et al. (2014). Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 59: 318–327.
Woo H-J, Reifman J . (2012). A quantitative quasispecies theory-based model of virus escape mutation under immune selection. Proc Natl Acad Sci USA 109: 12980–12985.
Wood GR, Ryabov EV, Fannon JM, Moore JD, Evans DJ, Burroughs N . (2014). MosaicSolver: a tool for determining recombinants of viral genomes from pileup data. Nucl Acids Res 42: e123.
Xia X, Xie Z, Salemi M, Chen L, Wang Y . (2003). An index of substitution saturation and its application. Mol Phylogenet Evol 26: 1–7.
Yang X, Charlebois P, Gnerre S, Coole MG, Lennon NJ, Levin JZ et al. (2012). De novo assembly of highly diverse viral populations. BMC Genomics 13: 475.
Zioni N, Soroker V, Chejanovsky N . (2011). Replication of Varroa destructor virus 1 (VDV-1) and a Varroa destructor virus 1–deformed wing virus recombinant (VDV-1–DWV) in the head of the honey bee. Virology 417: 106–112.
Acknowledgements
We thank the CB Dennis British Beekeepers' Research Trust for funding this research. GJM is funded by the British Beekeepers Association and the University of Reading. LW is funded by the Royal Society Dorothy Hodgkin Fellowship. SJM is funded by CB. Dennis, Apis._M and OECD. IMJ is funded by the UK Biotechnology and Sciences Research Council. DCS is funded by The Marine Biological Association Senior Research Fellowship. We thank Paul and Regula Schmid-Hempel for access to their archival bumblebee samples. Finally, we thank the sequencing services of University of Exeter and TGAC for their constructive suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Mordecai, G., Wilfert, L., Martin, S. et al. Diversity in a honey bee pathogen: first report of a third master variant of the Deformed Wing Virus quasispecies. ISME J 10, 1264–1273 (2016). https://doi.org/10.1038/ismej.2015.178
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.178
This article is cited by
-
Transmission of deformed wing virus between Varroa destructor foundresses, mite offspring and infested honey bees
Parasites & Vectors (2022)
-
Cold case: The disappearance of Egypt bee virus, a fourth distinct master strain of deformed wing virus linked to honeybee mortality in 1970’s Egypt
Virology Journal (2022)
-
First come, first served: superinfection exclusion in Deformed wing virus is dependent upon sequence identity and not the order of virus acquisition
The ISME Journal (2021)
-
Adapted tolerance to virus infections in four geographically distinct Varroa destructor-resistant honeybee populations
Scientific Reports (2021)
-
Transcriptome-level assessment of the impact of deformed wing virus on honey bee larvae
Scientific Reports (2021)

{kind=link}
{kind=link}
{kind=link}