
Abstract
The extent to which non-host-associated bacterial communities exhibit small-scale biogeographic patterns in their distribution remains unclear. Our investigation of biogeography in bacterial community composition and function compared samples collected across a smaller spatial scale than most previous studies conducted in freshwater. Using a grid-based sampling design, we abstracted 100+ samples located between 3.5 and 60 m apart within each of three alpine ponds. For every sample, variability in bacterial community composition was monitored using a DNA-fingerprinting methodology (automated ribosomal intergenic spacer analysis) whereas differences in bacterial community function (that is, carbon substrate utilisation patterns) were recorded from Biolog Ecoplates. The exact spatial position and dominant physicochemical conditions (for example, pH and temperature) were simultaneously recorded for each sample location. We assessed spatial differences in bacterial community composition and function within each pond and found that, on average, community composition or function differed significantly when comparing samples located >20 m apart within any pond. Variance partitioning revealed that purely spatial variation accounted for more of the observed variability in both bacterial community composition and function (range: 24–38% and 17–39%) than the combination of purely environmental variation and spatially structured environmental variation (range: 17–32% and 15–20%). Clear spatial patterns in bacterial community composition, but not function were observed within ponds. We therefore suggest that some of the observed variation in bacterial community composition is functionally ‘redundant’. We confirm that distinct bacterial communities are present across unexpectedly small spatial scales suggesting that populations separated by distances of >20 m may be dispersal limited, even within the highly continuous environment of lentic water.
Similar content being viewed by others


Introduction
Microbial communities constitute the majority of the Earth’s biomass and catalyse many ecosystem services, which are essential for sustaining life. For this reason, understanding the mechanisms that regulate the diversity and distribution of microorganisms, and their key functional traits, remains a topic of great interest and ecological importance. Free-living (that is, non-host-associated) bacteria have been shown to exhibit the capacity for dispersal on major oceanic and atmospheric currents resulting in ubiquitous distributions of many taxa (Morris et al., 2002; Ivars-Martinez et al., 2008). Despite their potential for global dispersal, it is evident that bacterial communities nevertheless exhibit clear biogeographic trends as distance–decay gradients in bacterial community similarity have been reported across a wide variety of landscapes and across a broad range of spatial scales (Horner-Devine et al., 2004; Oakley et al., 2010; Martiny et al., 2011; Lear et al., 2013). To date, however, most studies of bacterial biogeography compare samples collected >1 km apart (Fuhrman et al., 2008; Amend et al., 2012; Hanson et al., 2012; Monroy et al., 2012) and from a diverse range of ‘island-like’ habitats (Bell et al., 2005; Langenheder and Ragnarsson, 2007; Sommaruga and Casamayor, 2009; Bell, 2010), which are often known to exhibit substantial variability in environmental conditions. A major research question therefore remains to identify the distances at which patterns in microbial community composition and diversity manifest, and particularly the minimum spatial scales that significant biogeographic patterns can be detected.
Where observed, two main mechanisms are thought to be responsible for the distance–decay in compositional similarity in biological communities. First, if individuals are dispersal limited, we expect a decrease in community similarity with distance as individuals are more likely to colonise nearby locations. Second, as environmental attributes become less similar with distance, adaptation to local conditions, that is, ‘species sorting’, may also lead to a distance-decay in similarity. Recently, several studies have sought to disentangle the relative roles of these ecological mechanisms by analysing the strength of the relationship between community similarity and distance once environmental factors are accounted for, and vice versa (Beisner et al., 2006; Langenheder and Ragnarsson, 2007; Van der Gucht et al., 2007; Lear et al., 2013). Unexpectedly, evidence indicates that dispersal limitation may be more important in determining bacterial community composition and β-diversity at local scales than at larger regional or cross-continental scales. For example, a study of the composition of ammonia-oxidising bacteria in salt-marshes suggested that local-scale patchiness in community composition was caused by stochastic births and deaths, then accentuated by dispersal limitation of taxa over ecological timescales (Martiny et al., 2011). Alternatively, at the smallest spatial scales, high levels of immigration by poorly adapted taxa may override the effects of species sorting and ecological drift, thereby weakening the relationship between microbial community similarity and small-scale variability in environmental conditions. In such situations, community similarity may be more influenced by sample proximity to immigration sources. Here we investigate the importance of both geographic location and environmental factors in determining the composition of planktonic bacterial communities within three freshwater ponds where substantial and continual exchange of individuals is expected to occur over short time periods. Specifically, we chose to use a grid-based sampling design to map spatial variability in bacterial community composition (characterised using a DNA-fingerprinting methodology) across only small distances (3.5–60 m) within these shallow bodies of freshwater. Using this extremely fine-scale sampling approach, we explore spatial variation and patterns of distance–decay while simultaneously informing on the most appropriate scale to monitor the microbial community attributes of lentic freshwater.
If variation in bacterial composition is observed within our freshwater study sites, does it really matter? This question is relevant because high levels of functional redundancy are proposed to exist among bacterial taxa, whereby compositionally distinct communities perform similar functions, such as the degradation or mineralisation of organic matter (Rousk et al., 2009). The relationships between microbial community composition, diversity and functional potential have been explored in several aquatic bacterial community studies (Langenheder et al., 2005; Szabo et al., 2007; Comte and del Giorgio, 2010; Peter et al., 2011). However, such studies are rarely undertaken in natural systems (but see Parnell et al., 2010; Jiang et al., 2012) and are often conducted as analyses of ‘island-like’ systems such as batch culture microcosms (Langenheder et al., 2005) where species richness is frequently lowered artificially (Peter et al., 2011; Ylla et al., 2013) or in poorly connected environments where the exchange of microbial taxa among microcosms is otherwise impeded. Thus, it remains unclear the extent to which microbial communities exhibit biogeography in taxonomic or functional attributes across small spatial scales within contiguous landscapes. To address this gap in the literature, we conducted simultaneous carbon substrate-fingerprinting analysis of the communities at each sampling location within our ponds, thereby providing a measure of the changing functional, or metabolic attributes within each study site.
Using bacterial community data collected from shallow pond water, we asked three questions. First, at what spatial scale does the composition of bacterial communities become significantly different? Second, how well correlated are spatial variability in bacterial community composition and function? The answers to these questions have important implications for our understanding of community assembly processes and the use of compositional attributes in characterising microbial community function. Third, how important are environmental factors in explaining variation in bacterial community characteristics within ponds? We compare the environmental and spatial components of community variation to generate hypotheses of the importance of dispersal limitation and species sorting at these extremely small spatial scales; evidence of a significant role for environmental factors would imply that distinct bacterial niches develop, even within the apparently contiguous environment of freshwater.
Materials and methods
Study location and sample collection

The study was conducted in Tekapo Scientific Reserve, a flat rolling area of terminal moraine with scattered kettlehole tarns (shallow ponds), approximately 2 km south-east of Tekapo Village, New Zealand. Three ponds of broadly similar size were chosen for microbial community analysis (surface areas were 2450, 2800 and 3900 m2 for ponds 1, 2 and 3, respectively; Figure 1) and were all shallow pans, with a maximum depth of only 50 cm. The shallow depth of each pond is important as numerous studies reveal the significance of depth in determining the composition of aquatic bacterial communities (Shade et al., 2012). Using a 7 × 7 m grid layout, we chose to extract 33, 35 and 53 water samples from ponds 1, 2 and 3, which were sampled on 10, 11 and 12 January 2012, respectively. An additional set of samples were abstracted from pond 3 using a smaller grid format of 3.5 × 3.5 m such that spatial variability in bacterial community composition and function could be observed across a smaller spatial scale. To minimise any disturbance to each pond during sample collection, we used an adjustable hoist system to gradually lower a length of flexible tubing to a depth of 5 cm at each sampling location. A portable petrol engine pump was used to transfer water from the sampling location to the bankside for collection. Sample water was either used to record the pH and conductivity of the water using portable probes, stored at 4 °C for imminent microbial analysis or frozen at −20 °C for later chemical analysis. Following the abstraction of all sample water, the water temperature and concentration of dissolved oxygen were monitored in situ at every sampling location using a portable probe (550A YSI, Yellow Springs, OH, USA).

Map of sampling locations within Tekapo Scientific Reserve, Tekapo, New Zealand (41o01′04′′ S, 170o29′32′′ E). (a) Location of ponds sampled within the reserve. The shaded zones reveal the total area constrained by the shallow banks of each pond. Solid black lines reveal the presence of ephemeral channels for the flow of surface water. Water samples were collected from ponds 1, 2 and 3; (b–d) location of samples collected from ponds 1, 2 and 3, respectively. Most samples () were collected using a 7 × 7 m grid format. In pond 3, and additional 15 samples (•) were collected in three locations using a 3.5 × 3.5 m grid format (see inset, shaded). The solid line shows the location of pond banks; the dotted line shows the edge of the water on the day of sampling. Arrows show the direction of an ephemeral flow of water across the pond. (*) sample for which insufficient DNA was extracted for microbial community analysis. A full colour version of this figure is available at The ISMEJ Journal online.
Physicochemical analysis of water samples
Stored samples were analysed by high-performance liquid chromatography for concentrations of chloride, bromide, nitrate-N, nitrite-N, phosphate-P and sulphate-S, by analytical services at Lincoln University. Concentrations of both organic and inorganic carbon were assessed using a total carbon analyser (TOC-5000A, Shimadzu Oceania Pty Ltd, Sydney, NSW, Australia).
Molecular analysis of bacterial community composition
To concentrate bacterial cells for molecular analysis, 100 ml of water from each sample was passed through a Millipore Express PLUS polyethersulphone membrane filter (0.22 μm pore, 47 mm diameter; Merck Millipore, Eschborn, Germany) by vacuum filtration. DNA was then extracted from the membrane filter using a modified bead-beater method (Miller et al., 1999) to destroy the filter and lyse bacterial cells. We used the filter papers in replacement of 0.25 g soil in this method, which combines a bead-beating methodology with chloroform-isoamyl alcohol extraction, and precipitation of the extracted DNA with isopropanol. The bacterial community composition within every sample was analysed using automated ribosomal intergenic spacer analysis (or ARISA) following the method of (Lear et al., 2009) with the primers LDBact (5′-CCGGGTTTCCCCATTCGG-3′) and SDBact (5′-TGCGGCTGGATCCCCTCCTTC-3′) (Ranjard et al., 2001). The protocol of Ramette (2009) was used to identify ‘true peaks’ from ARISA electropherograms (that is, removing background noise generated during automated analysis) and bin fragments of similar size (using a selection window of 2 bp with a shift size of 0.1 bp). Fragments <150 or >1000 bp were considered to be comprised of PCR artefacts (for example, primer dimers) and therefore excluded from analysis. Each sample therefore consisted of 850 variables, hereafter referred to as operational taxonomic units (OTUs) in our study, which represent the length (in bp) of the intergenic spacer region of constituent bacteria. The total area of binned ARISA peaks were finally normalised (to 100) to removed differences in profiles caused by different initial DNA template quantities, and allow the relative abundance of these bacterial OTUs to be compared among samples.
Biolog substrate utilisation analysis of bacterial community function
Biolog EcoPlate’s (Hayward, CA, USA) were used to assess the carbon assimilation potential of microbial communities. Undiluted water (120 μl) from each water sample was added to each well of a different Biolog EcoPlate containing 31 different carbon sources (and a control well with no carbon source). Plates were incubated in darkness, at room temperature, and the absorbance of each well at 590 nm measured using an optical density microplate reader (BMG LABTECH FLUOstar Omega, Alphatech Systems Ltd, Auckland, New Zealand) every 24 h for 7 days. The optical density for each well was calculated as the substrate OD590 nm minus the control well OD590 nm. The data presented in this study were obtained after 48-h incubation when the majority of wells contained microbial communities in a state of exponential growth.
Statistical analyses
The similarity in bacterial community composition (ARISA data) or carbon substrate utilisation (Biolog data) among samples was documented using a Bray–Curtis similarity measure. To visualise multivariate patterns in bacterial community data, multidimensional scaling of each data set was done in the PRIMER v.6 computer program (Primer-E Ltd, Plymouth, UK) selecting a minimum stress value of 0.01 and 9999 restarts of the ordination data. To test for statistically significant variance among data sets (for example, comparing the community data collected from ponds 1, 2 and 3), permutational multivariate analyses of variance (PERMANOVA) were completed by comparing Bray–Curtis distances among sample profiles using an ‘add-on’ package for PRIMER, PERMANOVA+ (Anderson et al., 2008). In addition, from the PERMANOVA partitioning, direct multivariate analogues to the usual univariate analysis of variance estimators for variance components (Searle et al., 1992) were used to quantify the variability associated with each source of variation in the model. These are expressed in terms of their square-root (that is, as a pseudo‘s.d.’) in order to be on the same measurement scale as the original Bray–Curtis similarity measure, and are hereafter referred to simply as ‘sq. root var.’ values. To assess how closely related any two sets of multivariate data were (that is, comparing Bray–Curtis similarity matrices of the ARISA and Biolog data), Spearman’s ρ rank correlation coefficient was calculated via seriation with 9999 permutations of the data using the PRIMER RELATE routine.
As a small number of our sample sites had no bacterial OTUs in common, we plotted distance–decay relationships between bacterial community composition or function, and geographic distance, using the generalised dissimilarity model of (Millar et al., 2011). As we detected significant spatial variability in bacterial communities, we also chose to determine an optimal sampling intensity for alpine ponds based on this study data using a multivariate Mantel correlogram approach (the ‘mantel.correlog’ function in the R package ‘vegan’; Oksanen et al., 2012). A ‘holm’ correction for multiple testing was applied to protect the type I error rate (Legendre and Legendre, 2012).
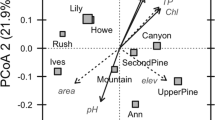
To visualise two-dimensional trends in bacterial community similarly across each pond, Bray–Curtis similarity data were subjected to a data reduction procedure, which used the best 1-day configuration scores for multidimensional scaling plots to divide the bacterial community data into 10n equally sized categories. Contour plots of bacterial community similarity (in composition or function across each pond) were then constructed displaying these groupings using the inverse distance weighting procedure in ArcMap v 10 (Environmental Systems Research Institute, Redlands, CA, USA). A similar approach was used to map spatial variability in environmental parameters across each pond. In this instance, the environmental data obtained from each sample were subjected to a data reduction procedure using principal components analysis performed in R version 3.0 (R_Core_Team, 2012). Separate contour plots, using 10 equally sized contour categories based on the principal components analysis sample scores that were interpreted using the inverse distance weighting procedure as above, were created for each pond and for each of the first three principal components of the environmental data (that is, PC1, PC2 and PC3).
Distance-based linear modelling using distance-based redundancy analysis of the Bray–Curtis distances among samples was performed in PERMANOVA+ for PRIMER to investigate the relationship between the ARISA or functional data, with both spatial and environmental variables recorded across the study sites. To test the significance of the relationship between each explanatory variable and the multivariate response variables, distance-based linear modelling (McArdle and Anderson, 2001) was built using forward selection and 9999 permutations of the data. Forward selection adds one variable, the variable that improves the selection criterion (R2) the most, at each step until no improvement in the selection criterion is possible. Using these R2 values, variance partitioning (Borcard et al., 1992) was performed with data from the distance-based linear modelling procedure to determine the proportion of variation in ARISA and functional data explained by each set of explanatory variables, namely (i) spatial variables, that is, geographic location data in the form of a full trend surface regression (Legendre and Legendre, 2012) including the data: easting (E), northing (N), E2, N2, EN, E2N, N2E, E3, N3; (ii) environmental variables, for example, pH and concentrations of total carbon, chloride, nitrite-N, nitrate-N, phosphate P, sulphate S; (iii) a combination of spatial and environmental factors or (iv) unexplained variance.
A limitation of this study was that it was not feasible to monitor spatial variability in the flow rate of water, which would most likely require spiking these protected status water bodies with chemical tracers. As a proxy for the flow rate of water, we chose to use the data of Tanentzap et al. (2013) to map fine-scale variability in plant species richness and the topology of ponds, which they monitored at 50 cm intervals across eight transects (data not shown). These measures were chosen for comparison as one would expect (i) water retention time to be greater in areas of the ponds where macrophytes are present to reduce the flow rate of water, and (ii) the flow rate of water to be greater where water is contained in ‘valleys’ between shallower sections of the pond bed, indicating a possible preferential flow path of water down the wider elevation gradient.
Results
On average, the compositional and functional attributes of bacterial communities varied between each pond (pairwise PERMANOVAs, all P<0.01) but with more of the variation in bacterial community composition being associated with pond identity (that is, ponds 1, 2 or 3) as compared with variation in bacterial community function (sq. root var. values were 37% and 11% for composition and function, respectively). Pairwise comparisons of average Bray–Curtis distances revealed the least similarity in bacterial community composition and function comparing sample data from ponds 1 and 2. A maximum of 101 different ARISA peaks (or bacterial OTUs) were detected in any single sample. However, taxa–area relationships (data not shown) revealed a plateau in bacterial OTU richness, reaching a maximum of ca 250 OTUs detected within any one pond. The total number of OTUs detected in this study was 335. A maximum of 153 OTUs were common to all three ponds, whereas 107 OTUs could only be detected in two ponds and a further 75 OTUs were detected in only one of the three ponds studied.
Distance-decay plots show that, although pairwise compositional distances between samples within ponds were highly variable, they declined significantly (all P<0.05) with geographic distance (Figure 2; Supplementary Figure S1). In contrast, pairwise functional distances between samples remained similar irrespective of distance (P=0.73, 0.53 and 0.39 for sample data collected within ponds 1, 2 and 3, respectively). Multivariate Mantel correlograms showed that for all ponds, pairwise comparisons of bacterial community composition became autocorrelated at a distance of less than approximately 20 m (that is, average bacterial community composition was significantly different comparing any samples located >20 m apart; Figure 3). Pairwise comparisons of bacterial community function were autocorrelated at a distance of ca 16 to 18 m in ponds 2 and 3; no significant autocorrelation was detected at any scale for pond 1.

Distance–decay relationship for bacterial community (a) composition and (b) function for pond 3. Each data point represents the Bray–Curtis similarity score for two samples and the geographic distance between the samples. Geographic distance varied between 3.5 and 62 m in pond 3. For each plot, the red line represents the predicted similarity based on a binomial generalised linear model, fitted using the approach of Millar et al. (2011). Data for ponds 1 and 2, and collectively comparing samples from all ponds are presented in Supplementary Figure S1. A full colour version of this figure is available at The ISMEJ Journal online.

Multivariate Mantel correlograms showing the significance of spatial autocorrelation in bacterial composition and function for grid sampling in three ponds at a resolution of 7 × 7 m. Solid black points represent scales with statistically significant spatial autocorrelation (positive Mantel correlation values) or spatial clustering (negative Mantel correlation values). Open points represent nonsignificant values. Holm’s correction was applied for multiple comparisons. Therefore, ‘scale values’ on the plots provide the approximate distances on the correlograms where spatial autocorrelation in bacterial composition or function between samples becomes nonsignificant (that is, only communities separated by distances greater than the scale values are likely differ significantly). These distances are slightly higher (more conservative) than reality because significance was tested for a series of classes (indicated by points on each graph).
Contour plots of the bacterial community data provide a visual representation of the nature and extent of bacterial community variability within each pond (Figure 4). In pond 1, different bacterial communities were present in the centre as compared with at the fringes of the water body. In contrast, differences in bacterial community composition varied across a NE/SW gradient for pond 2 and across a NW/SE gradient for pond 3. Therefore, although clear spatial patterns in bacterial community composition were observed, the nature of this trend differed within each pond. One, six and three OTUs were found to be present at every sampling location within ponds 1, 2 and 3, respectively; 10, 22 and 23 OTUs were present in ⩾75% of sampling locations within each pond. Spatial patterns in bacterial community function were not as pronounced as spatial patterns in bacterial community composition and no evidence was observed of any significant correlation between community composition and function within each pond (Spearman rank correlations, all P>0.05). A significant relationship between bacterial community composition and function was detected comparing data collected among the ponds (P=0.01); the relationship was weak, however, (ρ=0.114). The data of Tanentzap et al. (2013) provided no evidence of any clear relationship between bacterial community composition or function with pond depth. For example, Tanentzap et al. (2013) determined that the deepest section of pond 3 was located in the centre, whereas bacterial community composition varied across a clear NW/SE gradient. No flow of water was visible across the site suggesting that the movement of water was very slow during the time of study. However, a possible correlation between bacterial community composition with macrophyte species presence and richness was identified. For example, fewest macrophyte species were present in areas of pond 3 containing the bacterial communities depicted in light yellow in Figure 4, with the majority of samples in this area (that is, 17 out of 22) containing no macrophytes.

Similarity in bacterial community composition and function within ponds. Bacterial community data for each pond were subjected to a data reduction procedure by non-metric multidimensional scaling of Bray–Curtis similarity data. The difference between the highest and lowest one-dimensional (1D) configuration scores for each plot was used to divide the configuration into 10 equally sized categories. The bacterial community data falling within each of categories was assigned a different colour, across a gradient from yellow (lowest 1D configuration score) to dark blue (highest 1D configuration score) and plotted as a 2D-contour map using an inverse distance weighting procedure. The outcome of this approach is that samples hosting more similar bacterial community data are represented by more similar colours on each map. The 2D stress values for each multidimensional scaling plot used to plot the gradient data are shown in the bottom right of each plot.
Significant differences in environmental data among ponds (Supplementary Table S1) meant that more of the inter-pond variability in bacterial community composition and function were described by spatially structured variability in environmental factors, as compared with intra-pond variability. Within each pond, more of the variability in bacterial community composition (range: 50–70%) was explained by the spatial and environmental variables, as compared with variability in bacterial community function (range: 34–54%, Figure 5). Although very different patterns in bacterial community composition were observed in each pond, purely spatial variation always accounted for more of the variability in both bacterial community composition and function (ranges: 24–38% and 17–39%) than did environmental variation, or spatially structured environmental variation (ranges of the combined effects of environmental variation, and spatially structured environmental variation: 27–32% and 15–20%). Several of the environmental variables recorded in this study showed distinct spatial structuring within each pond (Supplementary Figure S2; Supplementary Table S2). However, as confirmed by variance partitioning (that is, by the percentage of ‘spatially structured environmental variation’), the relationship between spatial variability in environmental data with variability in bacterial community composition or function was weak (Figure 5). The only environmental variable to be consistently related to variability in bacterial community composition was concentrations of total carbon. However, the proportion of variation explained remained relatively low (5–15% of the total variation; Table 1), such that spatial patterns in bacterial composition and carbon nevertheless remain poorly comparable. In addition, weak but significant relationships between bacterial community composition and nitrite-N and phosphate-P were detected for pond 1 and pH for pond 3. No environmental variables were identified as being significantly related to variation in community function in any of the three ponds.

Results of partial regression analysis, partitioning the variation in bacterial community composition or function within each of the three ponds (data for each pond analysed individually). Four different components are shown: (hatched) unexplained variation; (black) pure environmental variation, independent of any spatial factors; (white) pure spatial variation, independent of any environmental factors; (grey) variation attributable to a combination of spatial and environmental factors.
Discussion
It is evident that bacterial communities exhibit clear biogeographic patterns in their distribution, with many studies reporting significant distance–decay in bacterial community composition (Langenheder and Ragnarsson, 2007; Soininen et al., 2007; Fuhrman et al., 2008; Bell, 2010; Martiny et al., 2011; Astorga et al., 2012; Lear et al., 2013). Nevertheless, few investigations have yet been conducted at fine (<1 km) spatial scales, and studies of lentic freshwater systems have typically relied on collecting only a small number of samples from each water body (but see Yannarell and Triplett, 2004 and Jones et al., 2012)). In contrast to most previous studies, our sampling protocol allowed the rapid collection of relatively large sample numbers across small areas, while also minimising site disturbance. Using these data, we confirm that distance–decay patterns occur in surface water bacterial communities and that community composition varies at smaller spatial scales than most previous studies have attempted to investigate (that is, <20 m). These findings could have significant implications for our understanding of spatial variability in microbial community dynamics and the appropriate scales for sampling microbial communities within aquatic landscapes. As a consequence of these findings, further study is now required so that we may determine if different bacterial communities are typically present at such fine scales within freshwater or if the spatial scaling of bacterial community characteristics varies widely among different lentic environments. This is of particular relevance since a study by Yannarell and Triplett (2004) previously identified no significant difference, on average, in bacterial community composition across scales of a few tens of metres in a range of larger waterbodies, with surface areas varying from 5000 to 39 000 000 m2. We consider it probable that the spatial scales at which patterns in microbial community characteristics manifest are reduced within small aquatic environments as they likely experience greater spatial variability in both environmental conditions and immigration (that is, being exposed to greater ‘edge-effects’ from the surrounding environment as compared with communities in the pelagic zone of large lakes or marine ecosystems).
The parameters that we measured explained 50–70% of the recorded variability in community similarity, despite (i) the low level of variability in various environmental characteristics detected across our study site, (ii) the lack of data for potentially important physicochemical variables such as concentrations of trace metals (Twiss and Campbel, 1998) and biotic variables, such as the abundance or composition of macroorganisms (Hanson et al., 2012), and (iii) the perceived limitations of DNA-fingerprinting (Blackwood et al., 2007), particularly the exclusion of rare taxa (Bent et al., 2007). In fact, the levels of unexplained variance reported here are lower than described for most aquatic microbial communities (Beisner et al., 2006; Van der Gucht et al., 2007; Lindstrom et al., 2010; Martiny et al., 2011). This may reflect the bias of previous studies to collect data from more isolated systems as the composition of disparate communities is more likely to be impacted by stochastic events such as random immigration or disturbance. Although such effects can significantly alter the evolutionary trajectory of poorly connected communities over time (Lee et al., 2013), the relative impacts may be reduced within more contiguous environments where the benefits and burdens provided by immigrant taxa may be more quickly distributed among the receiving community.
Although environmental factors explained a portion of the compositional variation, more variation was attributed to spatial factors, or geographic location. There are two possible explanations for this observation. First, we may not have measured some important spatially structured environmental factors that drive variation in bacterial composition. Second, rates of dispersal may have been low enough to allow bacterial communities to differentiate, via ecological drift, at a rate faster than they could be mixed together. Indeed, it is suggested that dispersal rates must be very high to cause the homogenisation of bacterial community composition by immigration (Logue and Lindstrom, 2010). It is likely that dispersal limitation did have an important role in determining the composition of bacterial communities among ponds as only 46% of the OTUs detected in this study were found to be present in all three ponds. Within ponds, however, the presumed mixing of water makes it less likely that dispersal limitation alone would be sufficient for pools of spatially segregated bacteria to develop. This is because mixing would result in community homogenisation by passive migration (Shade et al., 2010) as well as a reduction in environmental heterogeneity (Shade et al., 2008, 2010). Nevertheless, populations in small freshwater habitats are inevitably maintained by high intrinsic rates of immigration (also termed ‘mass effects’) from atmospheric deposition and the more populous sediment/water interface environments (Jiang et al., 2006). If large numbers of immigrant taxa are received by each pond, any preferential flow of water through the pond could be expected to increase spatial patterning in microbial community composition. This would result in more community variability being related to spatial variables that occur independently of any measured environmental variables, as observed in this study. Interestingly, the relative importance of spatial variables in determining bacterial community composition within each pond was greatest in the two ponds that are connected to other water bodies by ephemeral channels (for example, ponds 2 and 3; Supplementary Figures S1 and S2) during periods of high rainfall and therefore are also more likely to experience a greater rate and extent of immigration from neighbouring communities. Although it was not possible for us to map small-scale variability in the flow rate of water at each sampling location, it is perhaps inevitable that preferential flow paths of water do exist between, but also within each of these ponds. Of particular interest, the greatest variability in bacterial community composition within ponds was observed comparing areas with the most highest versus the lowest macrophyte species presence or richness. For example, sites in pond 3 containing no macrophytes were represented by a compositionally distinct group of bacteria (that is, shown as ‘yellow’ bacterial communities in Figure 4). Similarly, a greater abundance of macrophytes were present in the NE of pond 2 where compositionally distinct groups of bacteria were again observed. These data provide some evidence that spatial differences in bacterial community composition may be caused by spatial variation in the flow rate of water, which is known to be retarded by the presence of macrophytes in lentic freshwater (Madsen et al., 2001). In addition, however, differences in bacterial community composition could have been directly impacted by plant species abundance and composition, which are known to affect communities of phyllosphere-dwelling organisms in particular, and correlate with the nutritional qualities of plant exudates (Hempel et al., 2008). We recommend future studies of bacterial community composition in freshwater document macrophyte species density using analogous sampling techniques such that the relative importance of both spatial and physicochemical environmental factors on bacterial community attributes may be compared against local variability in macrophyte community attributes.
Although studies of the biogeographic distribution of various microbial phyla remain scarce, research on the biogeography of microbial functional traits remains particularly rare, with the exception of a number of experimental manipulations (Szabo et al., 2007; Rousk et al., 2010; Lindstrom and Ostman, 2011; Chaparro et al., 2012). In these naturally diverse freshwater communities, we found no evidence to suggest that the composition of a given bacterial community will predictably constrain it to performing certain biological functions. This is of particular interest given our choice of functional assay (C substrate utilisation) as concentrations of total C in the pond water were significantly related to spatial variability in bacterial community composition but not function. There are several possible reasons why we did not observe a distance–decay pattern in community substrate utilisation. First, the linkages between bacterial community composition and function may be weakened if non-bacterial members of the microbial communities were in fact responsible for significant carbon substrate utilisation. However, this is unlikely as the Biolog Ecoplate assay is tailored for bacterial community analysis and we found evidence only of very low numbers of fungi and protozoa in the Biolog culture plates. Second, the ‘culture bias’ introduced by the Biolog methods may have caused the assay to poorly reflect the functional diversity of the original microbial communities (Yin et al., 2000). Alternatively, the lack of relationship between bacterial community structure and function could provide additional support that the mass effects of immigration are an important driver of bacterial community composition within these ponds. We hypothesise this since many of the taxa that migrate into the ponds from neighbouring habitats will likely be poorly suited to the conditions in the pond water (for example, low nutrient levels) compared with the environment from which they departed (for example, the sediment water interface). If this is the case, large numbers of less functionally active, or even dormant taxa, may contribute to bacterial community composition, weakening the linkages between bacterial community composition and function (Lindstrom and Ostman, 2011). Of particular interest, a recent study by Severin et al. (2013) concluded that certain functional traits of bacterial communities were in fact driven more by the dispersion of cells than by habitat variation across short distances. Finally, it is likely that the outcomes of our study are impacted by our choice of functional assay as greater functional redundancy is expected to occur for coarse scale or generalist functions than functions, which require a high degree of specialisation (Nakatsu et al., 2005). It therefore remains likely that functionally specialist groupings of bacteria (for example, those capable of using a specific complex substrate as a sole source of carbon) would be more limited in their ability to disperse and colonise new habitats than more generalist groups of bacteria (for example, bacterial heterotrophs). Some evidence of this is provided in this study as a distance–decay in bacterial community composition was observed for bacterial communities growing on putrescine as a sole source of carbon, but not for communities growing on α-cyclodextrin or D-mannitol, which are potentially more simple carbon sources for degradation, being utilised by a larger number of bacterial taxa (data not shown). Regardless of the cause, we show that the functional attributes of culturable subsets of these microbial communities were poorly related to the biogeographic patterns displayed by the bacterial communities from which they originate.
In this study, we detected significant differences in bacterial community composition, but not function, within shallow freshwater ponds that were more closely related to variability in spatial location across the study site than to any recorded environmental variables. These observations may have significant implications for how we continue to sample freshwater microbial communities as significantly different bacterial communities were detected across distances of only 20 m. Additional studies, ideally using newly advanced molecular methods, should now be used to examine the extent of microbial functional biogeography within a broad range of freshwater habitats, and to inform on the likely importance of the observed variability in microbial community composition for the provision of a broad-range of key ecosystem services.
References
Amend AS, Oliver TA, Amaral-Zettler LA, Boetius A, Fuhrman JA, Horner-Devine MC et al. (2012). Macroecological patterns of marine bacteria on a global scale. J Biogeog 40: 800–811.
Anderson MJ, Gorley RN, Clarke KR . (2008) PERMANOVA+ for PRIMER: Guide to Software and Statistical Methods. PRIMER-E Ltd: Plymouth, UK.
Astorga A, Oksanen J, Luoto M, Soinen J, Virtanen R, Muotka T . (2012). Distance decay of similarity in freshwater communities: do macro- and microorganisms follow the same rules? Global Ecol Biogeogr 21: 365–375.
Beisner BE, Peres-Neto PR, Lindstrom ES, Barnett A, Longhi ML . (2006). The role of environmental and spatial processes in structuring lake communties from bacteria to fish. Ecology 87: 2985–2991.
Bell T . (2010). Experimental tests of the bacterial distance-decay relationship. ISME J 4: 1357–1365.
Bell T, Ager D, Song J-I, Newman JA, Thompson IP, Lilley AK et al. (2005). Larger islands house more bacterial taxa. Science 308: 1884.
Bent SJ, Pierson JD, Forney LJ . (2007). Measuring species richness based on microbial community fingerprints: the Emperor has no clothes. Appl Environ Microbiol 73: 2399–2401.
Blackwood CB, Hudleston D, Zak DR, Buyer JS . (2007). Interpreting ecological diversity indices applied to terminal restriction fragment length polymorphism data: insights from simulated microbial communities. Appl Environ Microbiol 73: 5276–5283.
Borcard D, Legendre P, Drapeau P . (1992). Partialling out the spatial component of ecological variation. Ecology 73: 1045–1055.
Chaparro JM, Sheflin AM, Manter DK, Vivanco JM . (2012). Manipulating the soil microbiome to increase soil health and plant fertility. Biol Fert Soil 48: 489–499.
Comte J, del Giorgio PA . (2010). Linking the patterns of change in composition and function in bacterioplankton successions along environmental gradients. Ecology 91: 1466–1476.
Fuhrman JA, Steele JA, Hewson I, Schwalbach MS, Brown MV, Green JL et al. (2008). A latitudinal diversity gradient in planktonic marine bacteria. Proc Natl Acad Sci USA 105: 7774–7778.
Hanson CJ, Fuhrman JA, Horner-Devine MC, Martiny JBH . (2012). Beyond biogeographic patterns: processes shaping the microbial landscape. Nat Rev Microbiol 10: 497–506.
Hempel M, Blume M, Blindow I, Gross EM . (2008). Epiphytic bacterila community composition on two submerged macrophytes in brackish water and freshwater. BMC Microbiol 8: 58.
Horner-Devine MC, Lage M, Hughes Martiny JB, Bohannan BJM . (2004). A taxa-area relationship for bacteria. Nature 432: 750–753.
Ivars-Martinez E, D'Auria G, Rodriguez-Valera F, Sanchez-Perez J-M, Ventosa A, Joint I et al. (2008). Biogeography of the ubiquitous marine bacterium Alteromonas macleodii determined by multilocus sequencing analysis. Mol Ecol 17: 4092–4106.
Jiang H, Dong H, Zhang G, Yu B, Chapman LR, Fields MW . (2006). Microbial diversity in water and sediment of Lake Chaka, an Athalassohaline Lake in Northwestern China. Appl Environ Microbiol 79: 3832–3845.
Jiang X, Langille MGI, Neches RY, Elliot M, Levin SA, Eisen JA et al. (2012). Functional biogeography of ocean microbes revealed through non-negative matrix factorization. PLoS One 7: e43866.
Jones SE, Cadkin TA, Newton RJ, McMahon KD . (2012). Spatial and temporal scales of aquatic bacterial beta diversity. Front Microbiol 3: 318 Article e318.
Langenheder S, Lindstrom ES, Tranvik LJ . (2005). Weak coupling between community composition and functioning of aquatic bacteria. Limnol Oceanogr 50: 957–967.
Langenheder S, Ragnarsson H . (2007). The role of environmental and spatial factors for the composition of aquatic and bacterial communities. Ecology 9: 2154–2161.
Lear G, Boothroyd IKG, Turner SJ, Roberts K, Lewis GD . (2009). A comparison of bacteria and benthic invertebrates as indicators of ecological health within streams. Freshwat Biol 54: 1532–1543.
Lear G, Washington V, Neale MW, Case B, Buckley HL, Lewis GD . (2013). The biogeography of stream bacteria. Global Ecol Biogeogr 22: 544–554.
Lee JE, Buckley HL, Etienne RS, Lear G . (2013). Both species sorting and neutral processes drive assembly of bacterial communities in aquatic microcosms. FEMS Microbiol Ecol 86: 288–302.
Legendre P, Legendre L . (2012) Numerical Ecology. Third English Edition. Elsevier: Amsterdam.
Lindstrom ES, Feng X-M, Graneli W, Kritzberg ES . (2010). The interplay between bacterial community composition and the environment determining function of inland water bacteria. Limnol Oceanogr 55: 2052–2060.
Lindstrom ES, Ostman O . (2011). The importance of dipersal for bacterial community composition and functioning. ISME J 6: e25883.
Logue JB, Lindstrom ES . (2010). Species sorting affects bacterioplankton community composition as determined by 16S rDNA and 16S rRNA fingerprints. ISME J 4: 729–738.
Madsen JD, Chambers PA, James WF, Koch EW, Westlake DF . (2001). The interaction between water movement, sediment dynamics and sumbersed macrophytes. Hydrobiologia 444: 71–84.
Martiny JBH, Eisen JA, Penn K, Allison SD, Horner-Devine MC . (2011). Drivers of bacterial beta-diversity depend on spatial scale. Proc Natl Acad Sci USA 108: 7850–7854.
McArdle BH, Anderson MJ . (2001). Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology 82: 290–297.
Millar RB, Anderson MJ, Tolimieri N . (2011). Much ado about nothings: using zero similarity points in distance-decay curves. Ecology 92: 1717–1722.
Miller DN, Bryant JE, Madsen EL, Ghiorse WC . (1999). Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl Environ Microbiol 65: 4715–4724.
Monroy F, Van der Putten WH, Yergeau E, Mortinmer SR, Duyts H, Bezemer TM . (2012). Community patterns of soil bacteria and nematodes in relation to geographic distance. Soil Biol Biochem 45: 1–7.
Morris RM, Rappe MS, Connon SA, Vergin KL, Siebold WA, Carlson CA et al. (2002). SAR11 clade dominates ocean surface bacterioplankton communties. Nature 420: 806–810.
Nakatsu CH, Carmosini N, Baldwin B, Beasley F, Kourtev P, Konopka A . (2005). Soil microbial community responses to additions of organic carbon substrates and heavy metals (Pb and Cr). Appl Environ Microbiol 71: 7679–7689.
Oakley BB, Carbonero F, Van der Gast CJ, Hawkins RJ, Purdy JK . (2010). Evolutionary divergence and biogeography of sympatric niche-differentiated bacterial populations. ISME J 4: 488–497.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB et al. (2012). Vegan: community ecology package. R package version 2.0-5 http://CRAN.R-project.org/package=vegan.
Parnell JJ, Rompato G, Latta LC, Pfrender ME, Van Nostrand JD, He Z et al. (2010). Functional biogeography as evidence of gene transfer in hypersaline microbial communities. PLoS One 5: e12919.
Peter H, Beier S, Bertilsson S, Lindstrom ES, Langenheder S, Tranvik LJ . (2011). Function-specific response to depletion of microbial diversity. ISME J 5: 351–361.
R_Core_Team (2012). R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna Austriahttp://www.R-projectorg.
Ramette A . (2009). Quantitative community fingerprinting methods for estimating the abundance of operational taxonomic units in natural microbial communities. Appl Environ Microbiol 75: 2495–2505.
Ranjard L, Poly F, Lata JC, Mougal C, Thioulouse J, Nazaret S . (2001). Characterization of bacterial and fungal soil communities by automated ribosomal intergenic spacer analysis fingerprints: biological and methodological variability. Appl Environ Microbiol 67: 4479–4487.
Rousk J, Baath E, Brookes PC, Lauber CL, Luzopone C, Caporaso JG et al. (2010). Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J 4: 1340–1351.
Rousk J, Brookes PC, Baath E . (2009). Contrasting soil pH effects on fungal and bacterial growth suggest functional redundancy in carbon mineralization. Appl Environ Microbiol 75: 1589–1596.
Searle SR, Casella G, McCulloch CE . (1992) Variance Components. John Wiley and Sons: New York.
Severin I, Ostman O, Lindstrom ES . (2013). Variable effects of dispersal on productivity of bacterial communities due to changes in functional trait composition. PLoS One 8: e80825.
Shade A, Chiu C-Y, McMahon KD . (2010). Seasonal and episodic lake mixing stimulate differential planktonic bacterial dynamics. Microbiol Ecol 59: 546–554.
Shade A, Jones SE, McMahon KD . (2008). The influence of habitat heterogeneity on freshwater bacterial commuinty composition and dynamics. Environ Microbiol 12: 455–466.
Shade A, Read JS, Youngblut ND, Fierer N, Knight R, Kratz TK et al. (2012). Lake microbial communities area resilient after a whole-ecosystem disturbance. ISME J 6: 2153–2167.
Soininen J, McDonald R, Hillebrand H . (2007). The distance decay of similarity in ecological communties. Ecography 30: 3–12.
Sommaruga R, Casamayor E . (2009). Bacterial 'cosmopolitanism' and importance of local environmental factors for community composition in remote high-altitude lakes. Freshwat Biol 54: 994–1005.
Szabo KE, Itor POB, Bertilsson S, Tranvik LJ . (2007). Importance of rare and abundant populations for the structure and functional potential of freshwater bacterial communities. Aquat Microb Ecol 47: 1–10.
Tanentzap AJ, Lee WG, Schulz KAC . (2013). Niches drive peaked and positive relationships between diversity and disturbance in natural ecosystems. Ecosphere 4: article 133.
Twiss MR, Campbel PGC . (1998). Trace metal cycling in the surface waters of Lake Erie: linking ecological and geochemical fates. J Great Lakes Res 24: 791–807.
Van der Gucht K, Cottenie K, Muylaert K, Vloemans N, Cousin S, Declerck S et al. (2007). The power of species sorting: local factors drive bacterial community composition over a wide range of spatial scales. Proc Natl Acad Sci USA 104: 20404–20409.
Yannarell AC, Triplett EW . (2004). Within- and between-lake variability in the composition of bacterioplankton communities: investigations using multiple spatial scales. Appl Environ Microbiol 70: 214–223.
Yin B, Crowley D, Sparovek G, De Melo WJ, Borneman J . (2000). Bacterial functional redundancy along a soil reclamation gradient. Appl Environ Microbiol 66: 4361–4365.
Ylla I, Peter H, Romani AM, Tranvik LJ . (2013). Different diversity-functioning relationship in lake and stream bacterial communities. FEMS Microbiol Ecol 85: 95–103.
Acknowledgements
This study was supported by RSNZ Marsden Fund (UOA0926). We acknowledge the kind assistance of Andrew Tanentzap and colleagues for the provision of plant species and water level data collected from Tekapo Scientific Reserve.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Lear, G., Bellamy, J., Case, B. et al. Fine-scale spatial patterns in bacterial community composition and function within freshwater ponds. ISME J 8, 1715–1726 (2014). https://doi.org/10.1038/ismej.2014.21
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.21
Keywords
This article is cited by
-
Molecular diversity and biogeography of benthic microeukaryotes in temperate seagrass (Zostera japonica) systems of northern China
Acta Oceanologica Sinica (2022)
-
Rare Bacteria Assembly in Soils Is Mainly Driven by Deterministic Processes
Microbial Ecology (2022)
-
Spatio-temporal dynamics of bacterial communities in the shoreline of Laurentian great Lake Erie and Lake St. Clair’s large freshwater ecosystems
BMC Microbiology (2021)
-
Seasonal dynamics of lotic bacterial communities assessed by 16S rRNA gene amplicon deep sequencing
Scientific Reports (2020)
-
Microbial Assemblage Dynamics Within the American Alligator Nesting Ecosystem: a Comparative Approach Across Ecological Scales
Microbial Ecology (2020)
