
Abstract
Viruses that infect the marine cyanobacterium Prochlorococcus have the potential to impact the growth, productivity, diversity and abundance of their hosts. In this study, changes in the microdiversity of cyanomyoviruses were investigated in 10 environmental samples taken along a North–South Atlantic Ocean transect using a myoviral-specific PCR-sequencing approach. Phylogenetic analyses of 630 viral g20 clones from this study, with 786 published g20 sequences, revealed that myoviral populations in the Atlantic Ocean had higher diversity than previously reported, with several novel putative g20 clades. Some of these clades were detected throughout the Atlantic Ocean. Multivariate statistical analyses did not reveal any significant correlations between myoviral diversity and environmental parameters, although myoviral diversity appeared to be lowest in samples collected from the north and south of the transect where Prochlorococcus diversity was also lowest. The results were correlated to the abundance and diversity of the co-occurring Prochlorococcus and Synechococcus populations, but revealed no significant correlations to either of the two potential host genera. This study provides evidence that cyanophages have extremely high and variable diversity and are distributed over large areas of the Atlantic Ocean.
Similar content being viewed by others


Introduction
Viruses are recognised as the most abundant biological entities in the oceans (Bergh et al., 1989; Suttle, 1994; Wommack and Colwell, 2000; Breitbart et al., 2002; Paul et al., 2002). Surface seawater typically contains 107 viral particles per ml, although concentrations of 1010 viral particles per ml have also been reported (Bergh et al., 1989; Jiang and Paul, 1994; Weinbauer et al., 1995; Wommack and Colwell, 2000). Viruses represent a major cause of microbial mortality in the oceans (Parada et al., 2008), and therefore have an important role in global biogeochemical cycling (Suttle, 2005, 2007) since lysis affects nutrient cycling and bacterial production (Wilhelm and Suttle, 1999; Mann, 2005; Suttle, 2005; Lennon et al., 2007). Viruses infecting the unicellular marine cyanobacterium Synechococcus were first isolated in 1993 (Suttle and Chan, 1993; Waterbury and Valois, 1993; Wilson et al., 1993). Most of these viruses, also called cyanophages, belong to the Myoviridae and are abundant and ubiquitous in the oceans. The diversity of these myoviruses has been studied in some detail using PCR primers specific for the major capsid protein g20 (Fuller et al., 1998; Wilson et al., 1999; Zhong et al., 2002; Sandaa and Larsen, 2006; Sullivan et al., 2006, 2008; Wilhelm et al., 2006).
Given their abundance, it has been postulated that cyanophages have the potential to significantly impact marine cyanobacterial assemblages (Suttle and Chan, 1993). For example, Mühling et al. (2005) analysed the diversity and abundance of both Synechococcus (using a rpoC1-based restriction fragment length polymorphism-approach; Mühling et al., 2006), and Synechococcus-infecting cyanophages (using g20 as molecular marker; Fuller et al., 1998), in the Gulf of Aqaba over an annual cycle and found that cyanophages had an important role, controlling of the diversity and abundance of the co-occurring Synechococcus population.
Cyanophages infecting Prochlorococcus have also been isolated and belong to three viral families: the Siphoviridae, Myoviridae and Podoviridae; the majority of cyanophage isolates belong to the latter two families (Sullivan et al., 2003, 2006). Although marine cyanophages have generally restricted host ranges, some have been shown not only to cross-infect the two major Prochlorococcus clades (low light (LL) and high light (HL)), but also to infect both LL Prochlorococcus and Synechococcus (Sullivan et al., 2003). The fact that cyanomyoviruses infect both groups of cyanobacteria means that it is not easy to use culture-independent approaches to distinguish between those cyanomyoviruses that infect only one of the two potential hosts and those that are capable of infecting both cyanobacterial genera.
Given the abundance of Prochlorococcus and its importance in the global biogeochemical cycle, we investigated the diversity of the cyanomyoviruses along a very long North–South Atlantic Ocean transect. Abundance and genetic diversity of Prochlorococcus and Synechococcus on this transect have been reported (Heywood et al., 2006; Zwirglmaier et al., 2007; Jameson et al., 2008, 2010). In this paper, we report simultaneous analysis (that is, parallel sample collection during the same cruise) of host and cyanophage, allowing us to assess the potential impact of these cyanomyoviruses on host populations.
Materials and methods
Sample collection
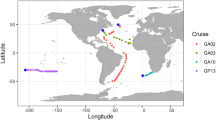
Sampling was carried out in September to October 2004 during the Atlantic Meridional Transect cruise (AMT-15) that sailed from Southampton (UK) to Cape Town (South Africa); details of this cruise have previously been described (Jameson et al., 2008). Ten samples have been selected from eight AMT-15 CTD (conductivity, temperature, depth sensor) sites (Figure 1) to study cyanomyovirus diversity; sample selection was primarily based on the known distribution of LL Prochlorococcus along the transect (Zwirglmaier et al., 2007; Jameson et al., 2008, 2010). Second, sampling sites were selected that were representative of different oceanic provinces sampled on the AMT, with a range of nutrient and productivity regimes. The eight stations selected for investigation (Figure 1) spanned a large geographic transect, from the northern temperate ocean (47° 92.34′ N, 14° 61.00′ W) to the southern boundary of the south Atlantic gyre (28° 58.02′ S, 06° 59.85′ W).

The AMT-15 cruise track with sampling stations indicated by corresponding CTD station numbers.
Seawater samples were filtered through 0.22-μm Sterivex cartridge filters (Millipore, Watford, UK) and the filtrate was used to concentrate the viral fraction. Two litres of the Sterivex filtrate were concentrated to 8–12 ml using a Vivaflow 200, with a 50 kDa molecular weight cut-off tangential flow unit (Sartorius, Epsom, UK). The tangential flow concentrate was transferred into a pre-conditioned dialysis membrane (14 kDa molecular weight cut-off; VWR, Lutterworth, UK). Dialysis membranes were immersed in the desiccant Polyethylene Glycol 8000 (PEG; Sigma-Aldrich, Poole, UK) for approximately 3–5 h at 4 °C after which the volume in the membrane had reduced to 2 ml. Viral concentrates were stored at −20 °C until subsequent analyses.
PCR amplification
The PCR amplifications were conducted as previously described (Wilson et al., 1999; Zhong et al., 2002; Sullivan et al., 2008). Before amplification, the viral concentrates were thawed and 1 μl was used as DNA template in a 25 μl PCR reaction. Cyanomyovirus g20 gene fragments were amplified by PCR using Taq polymerase (Invitrogen, Paisley, UK) and primer pair g20_CPS1.1 and g20_CPS8.1 (Sullivan et al., 2006). The following thermal cycle conditions were used: initial denaturation step at 96 °C for 1 min, followed by 35 cycles of 96 °C for 10 s, 35 °C for 30 s and 72 °C for 20 s, followed by a final extension step of 10 min at 72 °C. The presence of PCR products of the expected size range (541–550 base pairs) was confirmed on a 1% agarose gel. To compensate for potential biases in individual PCR reactions two independent PCRs were carried out for each sample and then combined before subsequent cloning. An aliquot of the PCR products from the pooled mix of the two independent PCRs was cloned into the pGEM-T vector system (Promega, Southampton, UK) following the manufacturer's instructions. Resultant individual colonies containing g20 DNA fragments were used for sequencing.
Sequencing
Double-stranded plasmid DNA containing the g20 fragments was sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Electrophoresis was carried out on an ABI 3100 automated sequencer (Applied Biosystems). The g20 sequences were added to the alignment of known cyanomyovirus g20 sequences in ARB (Ludwig et al., 2004). The final alignment encompassed the region of the g20 gene that was amplified with the primers used in this study (g20 fragments of 541–550 base pairs). Sequence data have been submitted to the EMBL/GenBank databases under the accession numbers FJ788950 to FJ789566. The clone names contain the following information: the prefix ‘g20’ denotes the sequence as a fragment of the g20 gene, the first number denotes the clone number, the second number represents the CTD sample number from the AMT-15 cruise, ‘14%’ and ‘1%’ refer to the percentage of surface irradiance observed at the sampling depth, and the final letters indicate the oceanic region (‘NG’ for northern gyre and ‘SG’ for southern gyre).
Phylogenetic analyses
Phylogenetic trees were constructed based on multiple amino acid alignments of g20 sequences constructed using MUSCLE and refined by GBlocks. Phylogenetic analyses were performed using the maximum likelihood algorithm, PhyML with aLRT to test for robustness by calculating approximated bootstrap values. Multiple phylogentic analyses with aLRT resulted in identical trees. Trees were rendered with FigTree. The amino acid sequences for cyanophage isolates and environmental clones used to construct the tree depicted by Sullivan et al. (2008) were obtained from the NCBI and the GOS databases (downloaded from http://camera.calit2.net/).
Statistical analyses
Multivariate statistical analyses were all carried out using PRIMER 6.0 (PRIMER-E, Plymouth, UK; Clarke et al., 2005). Each clone was assigned a phylotype derived from the phylogenetic analysis (Figure 2). Owing to variations in the number of positive clones obtained for each sampling site the data were normalised using the normalisation function in PRIMER 6.0. The g20 diversity was calculated for each sample in terms of richness and evenness using Margalef's species richness measure and using Simpson's index respectively. Ordination of samples was carried out to allow the relative similarity of cyanomyovirus populations from different samples to be compared. Patterns in the myoviral populations along the Atlantic transect were ordinated by comparison of the phylogenetic composition at the different sampling sites using non-parametric multidimensional scaling (MDS). For this, resemblance matrices were calculated on untransformed standardised data on the relative abundance of the g20 phylotypes in each clone library using Bray–Curtis similarity. The MDS was performed using the matrices with Kruskal's stress formula 1 at a minimum stress of 0.01 and with 50 restarts. The cyclicity and seriation of the MDS analysis was tested using the RELATE procedure. Cyclicity refers to a cyclical change starting with one population structure, transitioning through different populations and returning to an earlier population structure, while seriation describes a linear transition from one population structure to another. This procedure uses Spearman rank correlation to compare randomly permuted samples with the actual results, thus revealing the most highly correlated parameters (Clarke et al., 2005). Links between g20 phylogenetic clade relative abundance patterns and multivariate environmental factors were analysed using BIO-ENV within the BEST application. BEST is based on Spearman rank correlation between the two similarity matrices.

Cyanomyoviruses phylogenetic tree generated from a g20 amino acid alignment. The analysis contained two sequences from AMT-15 Prochlorococcus lysates, 786 sequences from previous studies and the 628 clones from the AMT-15 clone libraries. The enteric coliphage T4 was used to root the tree (first branch of clade I). Roman numerals at the circumference of the tree indicate new clade names, which contain sequences from previously described clades: I, heterotrophic phages: II, ‘novel clade I’ (Sullivan et al., 2008); III, ‘clade A’ (Zhong et al., 2002) and ‘novel clade II’ (Sullivan et al., 2008); IV, ‘clades B-F’ (Zhong et al., 2002) and some ‘novel clade I’ (Sullivan et al., 2008); V, mixed clades, this clade is followed by a small branch representing a single sequence; VIa, ‘novel clade III’ (Sullivan et al., 2008); VIb, ‘clade III’ (Zhong et al., 2002); VII, ‘clade I’ (Zhong et al., 2002); VIII–XI, redefined ‘clade II’ (Sullivan et al., 2008). Percentages on the branches represent bootstrap values for each named clade. Letters in parentheses after the clade number indicate phages isolated on Prochlorococcus (P) or Synechococcus (S).
Results
Environmental conditions at sampling stations
Sampling sites were chosen based on the overall abundance of Prochlorococcus measured by flow cytometry, and the abundance of the LL Prochlorococcus clade, which was determined using dot-blot hybridisation with DNA probes against the 16S ribosomal RNA molecule (Zwirglmaier et al., 2007). Analyses of the myoviral diversity focused largely, but not exclusively, on samples from the 1% light depth where LL Prochlorococcus cell numbers were at their highest; two additional samples (CTD stations 21, 68; Figure 1) were collected from shallower in the water column at the 14% light depth. Information on the abundance of Prochlorococcus and Synechococcus as well as total chlorophyll and biotic parameters at the sampling sites are summarised in Table 1. In essence, Prochlorococcus was the dominant cyanobacterium at all sampling sites—except CTD site 6 at the 1% light depth, where both Synechococcus and Prochlorococcus were present in approximately similar cell numbers. All of the viral samples were collected from within the upper surface mixed layer (above the thermocline), with the exception of the samples from CTD station 6 at the 1% light depth and CTD station 68 at the 14% light depth, where the samples were taken within the lower stratified layer.
Phylogenetic analyses
The viral capsid assembly protein g20 was used as the molecular marker to assess the genetic diversity of the myoviral populations in the environmental samples. However, the PCR amplifications did not always result in specific PCR products, despite stringent PCR conditions. This meant that the number of g20 fragments (after the exclusion of unspecific PCR fragments) recovered per clone library was variable (between 15 and 94 positive clones; Table 2). It is therefore unlikely that total saturation of g20 clades was reached for some sampling sites (for example, sampling site 6; Table 2). Sequence analyses of a total of 628 g20 clones from eight sampling stations with 10 viral DNA samples along the AMT-15 transect (Table 2) revealed that, within the examined gene fragment, the most variable region consisted of an insertion/deletion site, which varied between sequences by up to nine bases (that is, three amino acids).
Phylogenetic analyses were carried out based on an alignment consisting of these 628 AMT-15 g20 gene fragments, two further sequences from Prochlorococcus AMT-15 lysates and sequences from a further 786 g20 fragments from previously published studies (Zhong et al., 2002; Dorigo et al., 2004; Sullivan et al., 2008); (Figure 2). The addition of the g20 fragments from the whole genome sequences, and from other environmental studies, ensured that representative sequences included all g20 phylogenetic lineages identified to date. Overall, the phylogenetic analyses of these 1416 g20 sequences revealed that the g20 sequence clones obtained from the AMT-15 samples were distributed among all of the marine g20 clusters identified by previous studies, except for cluster IX that consisted of five environmental sequences (Figure 2; Supplementary Table S1). Moreover, the AMT-15 data resolved clade II—as identified by Sullivan et al. (2008)—into several subclades (Figure 2). Owing to the distinct deep branching pattern, these are now defined as new clades (IX, Xa, Xb, Xc and XI). Moreover, 96% of the sequences in clade Xc (Figure 2) were derived from the AMT-15 samples, thus representing a novel putative phylogenetic clade or subclade of a known phylogenetic lineage (that is, of clade II of Sullivan et al., 2008). The bootstrap values for each of the described clades varied between 67% and 91%, except for clade III, which had a low bootstrap value of 27% (Figure 2). Clade III is identical to that described by Sullivan et al. (2008), for which they gave no bootstrap value for comparison. The phylogenetic analyses also confirmed earlier findings that the g20 sequences from the myoviral isolates did not cluster according to the host (Synechococcus or Prochlorococcus) that was used for their isolation (Figure 2).
Changes in myoviral diversity along the Atlantic transect
The myoviral assemblages were analysed in terms of phylogenetic clade composition, based on comparison of the percentage of clones belonging to the various myoviral phylogenetic clades at each site (Figure 2). This required the data to be normalised against sampling efforts, thus compensating for the variable number of myoviral g20 clones analysed in each of the clone libraries (Supplementary Table S1; see above). This comparison confirmed that sequences clustering within clades X and XI were not only most prevalent in this study overall (Figure 2), but additionally, no sampling site was dominated by any clade other than clade X or XI (Figure 3). However, the comparison did not reveal an overall pattern of the composition of myoviral assemblages along the transect, although there was a certain degree of alternating dominance between clade XI and the most abundant subclades Xb and Xc (Figure 3). These three phylogenetic clusters represented between ca 70% and 90% of the clones in each clone library. The abundance of these three clusters changed along the north–south transect, with a single clade dominating at any one station. The exceptions were the N. Gyre stations 12 (1% light depth) 21 (14% light depth) and 68 (14% light depth), where dominance was split between Xb–XI and Xc–XI, respectively, comprising ca 70% of the total clones screened.

Histogram showing the percentage of g20 clones of each phylogenetic clade identified in Figure 2. Clade X is divided into phylogenetic subclusters. All samples were collected from the 1% light depth except for stations 21 (14%) and 68 (14%) which originated from the 14% light depth.
The lack of a statistically significant cyclicity or seriation (P=0.315 and P=0.061, respectively) of myoviral g20 diversity along the transect was further confirmed by MDS, based on the distribution of clones between phylogenetic clades (Figure 4). The MDS plot revealed similar myoviral assemblages at geographically remote sampling stations. The stations 21 (1% light depth) and 56 were indistinguishable and clustered with station 91. The N. Gyre station 21 (14% light depth) clustered closely to station 47 and of the S. Gyre station 68 (1% light depth). Additionally, the myoviral community of station 44 (1% light depth) and 68 (14% light depth) also clustered closely (Figure 3).

Nonparametric MDS ordination plot of myoviral diversity in the AMT-15 samples at the depths to which 1% of surface irradiation penetrated and the two samples (stations 21 and 68) from the 14% light depth. The analysis is based on Bray–Curtis similarities calculated from untransformed standardised data on the abundance of the myovirus g20 phylogenetic clades. The stress value (Kruskal's stress formula 1) of 0.12 indicates that the two-dimensional MDS plot is a good representation of the true distances in the multidimensional space. Relative distances between CTD samples reflect inter-sample similarities, with samples close together being similar in terms of their g20 phylogenetic clade composition. Arrows were added to assist interpretation, linking together samples by latitude, as they were collected north–south along the AMT-15 transect.
The myoviral diversity was further compared with abiotic environmental parameters (concentration of NO2−, NO3−, total N, PO43−, dissolved O2, temperature, depth) and the abundance of their potential cyanobacterial hosts. Quantitative information on the abundance of Prochlorococcus and Synechococcus clades was available for AMT-15, from studies by Jameson et al. (2010) and Zwirglmaier et al. (2007). BIO-ENV analyses (PRIMER 6.0) between the abiotic parameters and the myoviral g20 diversity revealed no correlations. For example, the BEST correlations between abiotic parameters and myoviral diversity (clade richness at the 1% light depths) were almost zero (r=0.09; P=0.20). In this context, it should be added that an r value of zero indicates no correlation, whilst an r value of 1 indicates perfect correlation. While the BEST correlation between myoviral diversity and Prochlorococcus diversity, specifically to the Prochlorococcus HLII clade, was higher (r=0.64), it was again not significant (P=0.47). Furthermore comparison of the myoviral diversity to the abundance of individual Prochlorococcus clades (HLII, HLI, LL, based on the rpoC1 gene) and Synechococcus (based on the 16S ribosomal RNA gene) phylogenetic clades, or Prochlorococcus rpoC1 restriction fragment length polymorphism-types, revealed no significant correlations of the abundance of Prochlorococcus HLII, HLI and LLI with myovirus richness and evenness (data not shown).
Discussion
Myoviral diversity
Myoviral diversity was high in the Atlantic Ocean and widely distributed, novel, putative phylogenetic clades have been revealed (Figures 2 and 4). The degree of diversity of g20 clones detected along the Atlantic transect was greater than previously found. There may be a number of explanations. In contrast to previous studies (Wilson et al., 2000; Lu et al., 2001; Zhong et al., 2002), a much larger number of clones (628) from different oceanic zones has been analysed in this study. Also, the g20 samples were collected from deeper in the water column than many previous studies (Wilson et al., 2000; Lu et al., 2001; Zhong et al., 2002; Sullivan et al., 2008). In addition, the highest diversity of cyanophages has been described at the deep chlorophyll maximum (Zhong et al., 2002), to which the 1% light depth correlates in this study (Table 1); at this depth, solar radiation is insufficient to cause significant viral decay (Suttle and Feng, 1992; Suttle and Chan, 1994).
The phylogenetic tree (Figure 2), combining the sequences obtained in this study (630) with those g20 sequences (786) analysed by Sullivan et al. (2008), resolves myoviral phylogeny based on g20 sequences. The branching pattern of the radial phylogenetic tree (Figure 2) is consistent with the tree of Zhong et al. (2002; ca 10% of the sequences) and also shows high similarity to the phylogenetic tree of Sullivan et al. (2008; ca 56% of the sequences; see also Supplementary Figure S1 for an alternate tree morphology). However, the tree presented here (Figure 2) reveals a much greater viral diversity. With the addition of the AMT-15 sequences from this study the radial tree indicates several individual clades and subclades; IX, Xa, Xb, Xc and XI (Figure 2) that comprise the previously described ‘culture-containing cluster II’, introduced by Sullivan et al. (2008). Indeed the deep branching pattern of ‘culture-containing cluster II’ was comparable to that seen between ‘culture-containing cluster I’ and ‘III’ and ‘novel cluster 2’ and ‘3’ combined (Sullivan et al., 2008). Figure 2 shows that our clades IX, Xa, Xb, Xc and XI form distinct branches and appear to represent novel putative clades. Xc was well represented in our samples and the clade consisted of 96% AMT-15 clones and no cultured isolates; likewise Xa contained no cultured isolates. These novel branches are unlikely to represent artefacts of the methodology and are supported by bootstrap values (68–89%), which are high given the large number of sequences. Further evidence for the robustness of the analysis comes from trees produced using the same method, but excluding the AMT-15 sequences. Comparable branching patterns were obtained to those previously documented (Zhong et al., 2002; Sullivan et al., 2008). The fact that clades Xa and Xc are well represented in the AMT-15 samples and contain no cultured isolates may be because of the stations and depths sampled, new emergent sequences and/or even the season of sampling.
The myoviral diversity varied over this North–South Atlantic Ocean transect, with the lowest myoviral g20 richness detected at the most northern and southern stations (Table 2). These stations also had lower Prochlorococcus diversity (Jameson et al., 2010) and were toward the limits of typical Prochlorococcus distribution. The survival of only a few dominant myoviral clades may have resulted from the corresponding reduced diversity and abundance of the Prochlorococcus host in these cooler waters at the extremities of the transect. In contrast, the greatest richness of g20 genetic clones was found at CTD station 44 (Table 2); this station was unremarkable in terms of Prochlorococcus abundance or diversity (Tables 1 and 2). Analyses of the whole data set showed no cyclicity in the g20 community structure, patterns seen north of the equator failed to reflect those south of the equator and there was no a strong linear progression from one community to another along the transect. Genetically similar cyanomyoviruses were geographically widely distributed with no apparent geographical segregation; this was also observed by Zhong et al. (2002) for open ocean waters. Nevertheless, very high viral g20 diversity was detected in the Atlantic Ocean, with significant variation of both community structure and diversity between sampling stations.
Cyanomyoviruses, environmental conditions and cyanobacterial hosts
The interdisciplinary nature of the AMT cruise series provides a wide range of ancillary data, including cyanobacterial abundance, diversity and environmental conditions for each sampling station. Therefore, a range of potentially influential factors could be investigated that might influence myoviral community structure and diversity. Cyanophages are subject to many external influences in the oceans, all of which may have an impact on their diversity: for example, host resistance/susceptibility (Waterbury and Valois, 1993), host abundance (Suttle and Chan, 1994), host starvation (Middelboe, 2000), ultraviolet degradation (Noble and Fuhrman, 1997), mixing (Murray and Jackson, 1993) and diurnal cycles (Clokie et al., 2006). However, this study that utilised a single ‘snapshot’ sampling approach that is characteristic of oceanic research cruises, revealed no significant correlations between cyanomyoviruses and any other measured parameter or combination of parameters. Furthermore, identical myoviral g20 clone sequences were detected at geographically distant sites, in different oceanic provinces with different nutrient status.
A number of explanations are possible for the absence of significant correlations. For example, Mühling et al. (2005) analysed an annual cycle from the same geographical sampling site in the Gulf of Aqaba and showed that myoviral diversity and abundance were correlated to the co-occurring Synechococcus population diversity and abundance; this correlation was even higher when a lag-time of 1 month was introduced. Such a lag between host and viral population dynamics was also observed by Bratbak et al. (1996) in coastal waters. Crucially, our study assessed host and viral diversity simultaneously—samples that were discrete in both time and space. A further factor that may help to explain the lack of correlation is that in surface waters, viral turnover rates are variable, in the order of hours (Bettarel et al., 2002; Clokie et al., 2006) to a few days (Parada et al., 2008). For these reasons, it may be unreasonable to expect that a single snapshot will reveal infection dynamics.
Another difficulty in detecting relationships between myoviruses and their hosts is that it is not possible to use g20 as the marker gene to distinguish between Prochlorococcus- and Synechococcus-infecting myoviruses. In fact, Sullivan et al. (2008) recently provided ample evidence that phage portal proteins are not good predictors of a phage's host or habitat. The complexity of these processes is further confirmed by the finding that g20 is part of a region of the myoviral genome that is highly mobile and is therefore often transferred between viruses (Monod et al., 1997). Moreover, Short and Suttle (2005) have previously indicated a need for caution in attributing all g20 sequences to cyanophages. However, the improved primers used in this study (g20_CPS1.1 and g20_CPS8.1) differed from those used by Short and Suttle (2005; CPS4 and G20-2), and have been empirically tested on a wide array of cyanophage isolates (Sullivan et al., 2008).
Conclusions
A large proportion of the AMT-15 g20 sequences fell into novel putative clades, which contained no cultured phage isolates (notably Xa and Xc, Figure 2). These findings are inconsistent with an assumption of Sullivan et al. (2008) that cyanophage with no cultured representatives are rare. In order to understand the role of these viral groups in the ecosystem and their impact on host populations it is important to continue to isolate phage from the environment. Viruses are dependent on host abundance and diversity and many cyanophage have demonstrated limited host ranges (Sullivan et al., 2003). However, in this study, host and viral dynamics were not correlated. Furthermore, this study raises questions over the value of g20 for discrete time-space samples, because the g20 myoviral marker did not appear to operate as a functional marker of phage-host specificity. This is consistent with the supposition of Sullivan et al. (2008) that the g20 protein and phage–host interactions are uncoupled evolutionarily. Therefore, the use of alternative marker genes with stronger links to host range, may have more value in elucidating these interactions; candidates could be viral-encoded host genes (Millard et al., 2004; Sullivan et al., 2006; Lindell et al., 2007) or viral genes involved in host recognition (Weigele et al., 2007; Chai et al., 2010). It has become clear that the assessment of viral–host dynamics from single discrete samples is difficult. To truly investigate viral–host dynamics, it may be necessary to evaluate both temporal and spatial dynamics of phage–host communities—a combination of Lagrangian and Eulerian sampling—in order to enhance our ability to predict the impacts of viruses on global ecosystem function. Although sampling constraints on this large oceanographic transect have not elucidated host–virus dynamics, this study has revealed extremely high g20 diversity and putatively novel clades with no spatial link to host abundance or diversity.
References
Bergh O, Borsheim KY, Bratbak G, Heldal M . (1989). High abundance of viruses found in aquatic environments. Nature 340: 467–468.
Bettarel Y, Dolan JR, Hornak K, Lemee R, Masin M, Pedrotti ML et al. (2002). Strong, weak, and missing links in a microbial community of the NW Mediterranean Sea. FEMS Microb Ecol 42: 451–462.
Bratbak G, Heldal M, Thingstad TF, Tuomi P . (1996). Dynamics of virus abundance in coastal seawater. FEMS Microb Ecol 19: 263–269.
Breitbart M, Salamon P, Andresen B, Mahaffy JM, Segall AM, Mead D et al. (2002). Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci USA 99: 14250–14255.
Chai YM, Xiong HY, Ma XY, Cheng LQ, Huang GR, Rao ZL et al. (2010). Molecular characterization, structural analysis and determination of host range of a novel bacteriophage LSB-1. Virology J 7: 255.
Clarke KR, Warwick RM, Gorley RN, Somerfield PJ . (2005). Change in Marine Communities: An Approach to Statistical Analysis and Interpretation, 3rd edn. Primer-E: Plymouth, UK.
Clokie MRJ, Millard AD, Mehta JY, Mann NH . (2006). Virus isolation studies suggest short-term variations in abundance in natural cyanophage populations of the Indian Ocean. J Mar Biol Asso 86: 499–505.
Dorigo U, Jacquet S, Humbert JF . (2004). Cyanophage diversity, inferred from g20 gene analyses, in the largest natural lake in France, Lake Bourget. Appl Environ Microbiol 70: 1017–1022.
Fuller NJ, Wilson WH, Joint IR, Mann NH . (1998). Occurrence of a sequence in marine cyanophages similar to that of T4 g20 and its application to PCR-based detection and quantification techniques. Appl Environ Microbiol 64: 2051–2060.
Heywood JL, Zubkov MV, Tarran GA, Fuchs BM, Holligan PM . (2006). Prokaryoplankton standing stocks in oligotrophic gyre and equatorial provinces of the Atlantic Ocean: evaluation of inter-annual variability. Deep Sea Res Part II 53: 1530–1547.
Jameson E, Joint I, Mann NH, Mühling M . (2008). Application of a novel rpoC1-RFLP approach reveals that marine Prochlorococcus populations in the Atlantic gyres are composed of greater microdiversity than previously described. Microb Ecol 55: 141–151.
Jameson E, Joint I, Mann NH, Mühling M . (2010). Detailed analysis of the microdiversity of Prochlorococcus populations along a North-South Atlantic Ocean transect. Environ Microbiol 12: 156–171.
Jiang SC, Paul JH . (1994). Seasonal and diel abundances of viruses and occurrence of lysogeny/bacteriocinogeny in the marine environment. Mar Ecol Prog Ser 104: 163–172.
Lennon JT, Khatana SAM, Marston MF, Martiny JBH . (2007). Is there a cost of virus resistance in marine cyanobacteria? ISME J 1: 300–312.
Lindell D, Jaffe JD, Coleman ML, Futschik ME, Axmann IM, Rector T et al. (2007). Genome-wide expression dynamics of a marine virus and host reveal features of co-evolution. Nature 449: 83–86.
Lu J, Chen F, Hodson RE . (2001). Distribution, isolation, host specificity, and diversity of cyanophages infecting marine Synechococcus spp. in river estuaries. Appl Environ Microbiol 67: 3285–3290.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar B et al. (2004). ARB: a software environment for sequence data. Nucl Acid Res 32: 1363–1371.
Mann NH . (2005). The third age of phage. PLoS Biol 3: 753–755.
Middelboe M . (2000). Bacterial growth rate and marine virus-host dynamics. Microb Ecol 40: 114–124.
Millard A, Clokie MRJ, Shub DA, Mann NH . (2004). Genetic organization of the psbAD region in phages infecting marine Synechococcus strains. Proc Natl Acad Sci USA 101: 11007–11012.
Monod C, Repoila F, Kutateladze M, Tetart F, Krisch HM . (1997). The genome of the pseudo T-even bacteriophages, a diverge group that resembles T4. J Mol Biol 267: 237–249.
Mühling M, Fuller NJ, Millard A, Somerfield PJ, Marie D, Wilson WH et al. (2005). Genetic diversity of marine Synechococcus and co-occurring cyanophage communities: evidence for viral control of phytoplankton. Environ Microbiol 7: 499–508.
Mühling M, Fuller NJ, Somerfield PJ, Post AF, Wilson WH, Scanlan DJ et al. (2006). High resolution genetic diversity studies of marine Synechococcus using rpoC1-based RFLP. Aquat Microb Ecol 45: 263–275.
Murray AG, Jackson GA . (1993). Viral dynamics II: a model of the interaction of ultraviolet light and mixing processes on virus survival in seawater. Mar Ecol Prog Ser 102: 105–114.
Noble RT, Fuhrman JA . (1997). Virus decay and its causes in coastal waters. Appl Environ Microbiol 63: 77–83.
Parada V, Baudoux AC, Sintes E, Weinbauer MG, Herndl GJ . (2008). Dynamics and diversity of newly produced virioplankton in the North Sea. ISME J 2: 924–936.
Paul JH, Sullivan MB, Segall AM, Rohwer F . (2002). Marine phage genomics. Comp Biochem Physiol B: Biochem Mol Biol 133: 463–476.
Sandaa RA, Larsen A . (2006). Seasonal variations in virus-host populations in Norwegian coastal waters: focusing on the cyanophage community infecting marine Synechococcus spp. Appl Environ Microbiol 72: 4610–4618.
Short CM, Suttle CA . (2005). Nearly identical bacteriophage structural gene sequences are widely distributed in both marine and freshwater environments. Appl Environ Microbiol 71: 480–486.
Sullivan MB, Coleman ML, Quinlivan V, Rosenkrantz JE, DeFrancesco AS, Tan G et al. (2008). Portal protein diversity and phage ecology. Environ Microbiol 10: 2810–2823.
Sullivan MB, Lindell D, Lee JA, Thompson LR, Bielawski JP, Chisholm SW . (2006). Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol 4: 1344–1357.
Sullivan MB, Waterbury JB, Chisholm SW . (2003). Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature 424: 1047–1051.
Suttle CA . (1994). The significance of viruses to mortality in aquatic microbial communities. Microb Ecol 28: 237–243.
Suttle CA . (2005). Viruses in the sea. Nature 437: 356–361.
Suttle CA . (2007). Marine viruses—major players in the global ecosystem. Nat Rev Microbiol 5: 801–812.
Suttle CA, Chan AM . (1993). Marine cyanophages infecting oceanic and coastal strains of Synechococcus—abundance, morphology, cross-infectivity and growth-characteristics. Mar Ecol Prog Ser 92: 99–109.
Suttle CA, Chan AM . (1994). Dynamics and distribution of cyanophages and their effect on marine Synechococcus spp. Appl Environ Microbiol 60: 3167–3174.
Suttle CA, Feng C . (1992). Mechanisms and rates of decay of marine viruses in seawater. Appl Environ Microbiol 58: 3721–3729.
Waterbury JB, Valois FW . (1993). Resistance to co-occurring phages enables marine Synechococcus communities to coexist with cyanophages abundant in seawater. Appl Environ Microbiol 59: 3393–3399.
Weigele PR, Pope WH, Pedulla ML, Houtz JM, Smith AL, Conway JF et al. (2007). Genomic and structural analysis of Syn9, a cyanophage infecting marine Prochlorococcus and Synechococcus. Environ Microbiol 9: 1675–1695.
Weinbauer MG, Fuks D, Pusharic S, Peduzzi P . (1995). Diel, seasonal, and depth-related variability of viruses and dissolved DNA in the Northern Adriatic sea. Microb Ecol 30: 25–41.
Wilhelm SW, Carberry MJ, Eldridge ML, Poorvin L, Saxton MA, Doblin MA . (2006). Marine and freshwater cyanophages in a Laurentian Great Lake: evidence from infectivity assays and molecular analyses of g20 genes. Appl Environ Microbiol 72: 4957–4963.
Wilhelm SW, Suttle CA . (1999). Viruses and nutrient cycles in the sea—viruses play critical roles in the structure and function of aquatic food webs. Bioscience 49: 781–788.
Wilson WH, Fuller NJ, Joint IR, Mann NH . (2000). Analysis of cyanophage diversity in the marine environment using denaturing gradient gel electrophoresi. In: Bell CR, Brylinsky M, Johnson-Green P (eds). Microbial Biosystems: New frontiers. Proceeding of the 8th International Symposium on Microbial Ecology. Atlantic Canada Society for Microbial Ecology. Atlantic Canada Society for Microbial Ecology: Halifax, Nova Scotia, Canada, pp 565–571.
Wilson WH, Joint IR, Carr NG, Mann NH . (1993). Isolation and molecular characterization of 5 marine cyanophages propagated on Synechococcus sp strain WH7803. Appl Environ Microbiol 59: 3736–3743.
Wilson WH, Nicholas JF, Joint IR, Mann NH . (1999). Analysis of cyanophage diversity and population structure in a south-north transect of the Atlantic Ocean. Bull Inst Oceanogr Fish 19: 209–216.
Wommack KE, Colwell RR . (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol Mol Biol Rev 64: 69–114.
Zhong Y, Chen F, Wilhelm SW, Poorvin L, Hodson RE . (2002). Phylogenetic diversity of marine cyanophage isolates and natural virus communities as revealed by sequences of viral capsid assembly protein gene g20. Appl Environ Microbiol 68: 1576–1584.
Zwirglmaier K, Heywood JL, Chamberlain K, Woodward EMS, Zubkov MV, Scanlan DJ . (2007). Basin-scale distribution patterns lineages in the Atlantic Ocean. Environ Microbiol 9: 1278–1290.
Acknowledgements
We thank the captain and crew of the RRS Discovery. In this study CTD/underway data from the Atlantic Meridional Transect Consortium (NER/0/5/2001/00680) was used, provided by the British Oceanographic Data Centre (BODC) and supported by the Natural Environment Research Council (NERC). This work was supported by a NERC research studentship (NER/S/A/2003/11883A) allocated to EJ. We thank Katie Chamberlain (PML) for the nutrient data. This is contribution number 180 of the AMT programme.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Jameson, E., Mann, N., Joint, I. et al. The diversity of cyanomyovirus populations along a North–South Atlantic Ocean transect. ISME J 5, 1713–1721 (2011). https://doi.org/10.1038/ismej.2011.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2011.54

{kind=link}