
Abstract
Myxobacteria are common in terrestrial habitats and well known for their formation of fruiting bodies and production of secondary metabolites. We studied a cluster of myxobacteria consisting only of sequences of marine origin (marine myxobacteria cluster, MMC) in sediments of the North Sea. Using a specific PCR, MMC sequences were detected in North Sea sediments down to 2.2 m depth, but not in the limnetic section of the Weser estuary and other freshwater habitats. In the water column, this cluster was only detected on aggregates up to a few meters above the sediment surface, but never in the fraction of free-living bacteria. A quantitative real-time PCR approach revealed that the MMC constituted up to 13% of total bacterial 16S rRNA genes in surface sediments of the North Sea. In a global survey, including sediments from the Mediterranean Sea, the Atlantic, Pacific and Indian Ocean and various climatic regions, the MMC was detected in most samples and to a water depth of 4300 m. Two fosmids of a library from sediment of the southern North Sea containing 16S rRNA genes affiliated with the MMC were sequenced. Both fosmids have a single unlinked 16S rRNA gene and no complete rRNA operon as found in most bacteria. No synteny to other myxobacterial genomes was found. The highest numbers of orthologues for both fosmids were assigned to Sorangium cellulosum and Haliangium ochraceum. Our results show that the MMC is an important and widely distributed but largely unknown component of marine sediment-associated bacterial communities.
Similar content being viewed by others


Introduction
Myxobacteria are unique, with a life style differing from all other prokaryotes. They are capable of excreting hydrolytic enzymes and decomposing various and complex biopolymers but can also lyse and degrade other prokaryotes and even eukaryotes (Shimkets et al., 2006). Myxobacterial cells can spread and swarm on an excreted polymeric, mucus-like matrix. Most notably, under unfavourable nutrient conditions, many myxobacteria can aggregate to dense mushroom-like cell colonies and produce fruiting bodies and spores (Shimkets, 1990; Shimkets et al., 2006). In this complex, density-dependent life style, signalling compounds have a key role (Goldman et al., 2006), and myxobacteria are well known for the production of various secondary metabolites and thus are prime targets for the search of new secondary metabolites (Gerth et al., 2003; Wenzel and Müller, 2007; Weissman and Müller, 2010). They contain the largest genomes of all prokaryotes and the genome of Sorangium cellulosum, with >13 Mbp as the largest bacterial genome sequenced to date (Schneiker et al., 2007). Myxobacteria form a phylogenetically coherent group and constitute the order Myxococcales in the class Deltaproteobacteria. They are subdivided into the three suborders Cystobacterineae, Sorangiineae and Nannocystineae (Reichenbach, 2005; Garcia et al., 2010).
Myxobacteria are widespread in terrestrial habitats. Historically and until recently, soil, dung and plant detritus were considered as the typical environments of these organisms (Dawid, 2000). Myxobacteria have been isolated also from limnetic habitats; however, they were considered as being washed in from the terrestrial surroundings and not as indigenous limnetic organisms (Reichenbach, 1999). For a long time, it was assumed that myxobacteria are not able to dwell in saline and marine habitats. Some myxobacteria were isolated from marine environments but exhibited a growth optimum at rather low salinities and, therefore, were thought to be salt tolerant and brought in from the terrestrial surroundings, comparable to those in freshwater habitats (Rückert, 1984). During the recent past, several myxobacteria have been isolated from marine habitats and shown to be able to grow at sodium chloride concentrations around that of sea water, for example, Haliangium ochraceum and H. tepidum (Foudou et al., 2002), Plesiocystis pacifica (Iizuka et al., 2003a) and Enhygromyxa salina (Iizuka et al., 2003b). All these species originate from coastal regions of Japan and affiliate to the suborder Nannocystineae. Halotolerant and halophilic myxobacteria appear to differ with respect to the formation of fruiting bodies, and the adaptation of their life cycle to the salty environment (Zhang et al., 2005). Selected strains of Myxococcus form fruiting bodies also in sea water media (Wang et al., 2007). Recently, Jiang et al. (2010) reported several clusters of myxobacterial sequences that were found in a few deep sea sediment samples and at one hydrothermal vent site. These myxobacteria are phylogenetically separated from their terrestrial counterparts, suggesting that distinct clusters of marine myxobacteria do exist. As nothing else than the phylogeny of these myxobacteria is known so far, further studies are needed to elucidate the ecology, physiology and genomics of these organisms.
During an investigation of the bacterial communities in the German Wadden Sea, southern North Sea, by denaturing gradient gel electrophoresis of PCR-amplified 16S rRNA gene fragments, we persistently detected a phylotype affiliated to the Myxococcales in the surface sediment (Stevens et al., 2005). This phylotype is related to several other uncultured phylotypes detected in various marine sediments, but only distantly to described species (<90% sequence similarity). Therefore, we comprehensively studied the occurrence of these myxobacteria in the North Sea and other marine systems. The study was complemented by analysis of a fosmid library from North Sea sediment. Two fosmids carrying 16S rRNA genes closely affiliated with other phylotypes of the new cluster were obtained and completely sequenced.
Materials and methods
Study sites and sampling
Sediment and water samples in the North Sea were taken at 12 stations during a cruise with RV Heincke in September 2005, covering a transect from Bremerhaven, Germany, to 58°N close to the Norwegian coast (Figure 1a, Supplementary Table 1). For DNA extraction, 250 ml of seawater was prefiltered onto 5.0 μm polycarbonate filters (Nuclepore, Whatman Schleicher Schuell, Piscataway, NJ, USA) to obtain the fraction of particle-associated bacteria and subsequently onto 0.2 μm polycarbonate filters to obtain that of free-living bacteria. Sediment from the surface (0–0.5 cm horizon) was taken using a multiple corer. Samples were immediately frozen after sampling and stored at −20 °C until further processing. Further sediment samples (0–0.5 cm horizon) for the detection of myxobacteria were obtained from different water depths and oceanic locations, including the Atlantic, Pacific, Indian and Arctic Ocean, the Baltic, Mediterranean and Black Sea, and hypersaline waters (Supplementary Table 1). Samples for DNA extraction and subsequent construction of a fosmid library were taken on 23 March 2002 from surface sediment of an intertidal sand flat (Janssand) in the German Wadden Sea (53°43 N, 07°41 E). The cores were sectioned and immediately frozen at −20 °C. DNA taken for library construction was extracted from sediment of the 5–12 cm horizon (see below). A detailed description of all sampling procedures is given in the Supplementary Text 1.

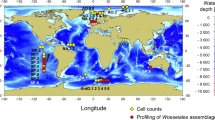
(a). Stations (St) in the North Sea visited during a cruise with RV Heincke in September 2005 and analysed for the presence of the MMC. (b). Abundance of 16S rRNA genes of the MMC (percent (%) of total bacterial 16S rRNA genes) in sediment surface samples of the various stations. Numbers on the y-axis refer to stations given in a.
Nucleic acid extraction
DNA for subsequent screening with a PCR specific for the marine myxobacteria cluster (MMC) was extracted following the protocol of Zhou and colleagues (1996), with modifications described by Giebel et al. (2009). DNA extraction was checked on a 1% agarose gel. Stock DNA was stored at −80 °C and subsamples at −20 °C until further analysis.
Design of the MMC PCR detection system
On the basis of available 16S rRNA gene sequences of the MMC, specific primers were designed using the ProbeDesign function of the ARB package (http://www.arb-home.de/). Two primer systems to detect the MMC were designed: MMC655f (5-AGTAATGGAGAGGGTGGC-3)/MMC841r (5-GGCACAGCAGAGGTCAAT-3) and MMC583f (5-AGGCGGACTCGCAAGTCG-3)/MMC734r (5-GTAAATGTCCAGGTGGC-3). Specificity of the primer sequences was checked in silico with the NCBI and RDP databases (http://www.ncbi.nlm.nih.gov/; http://rdp.cme.msu.edu/) and resulted in at least one mismatch to other organisms for MMC655f and MMC583f, and two mismatches for MMC734r. For the primer MMC841r, two sequences not included in the MMC showed no mismatch. As the first primer set covers more sequences of the MMC than the second, the first system was chosen for screening of environmental samples. The second MMC primer system was used for quantitative PCR because of higher specificity (see below). A cloned 16S rRNA gene fragment of a MMC bacterium was used as positive control and standard. Conditions for the MMC-specific PCR (primers MMC655f/MMC841r and MMC655f/1492r) and details on the development of the PCR conditions are given in the Supplementary Text 1. All DNA samples were prechecked with bacteria-specific primers 341f and 907r (Muyzer et al., 1998) before specific PCR.
Cloning and sequencing of PCR products
To prove that positive screening results obtained from PCR with the primer pair MMC655f/MMC841r and environmental samples derived from bacteria of the MMC, PCR products obtained with randomly selected samples and the combination of primer MMC655f and the bacteria-specific primer 1492r (Muyzer et al., 1995) were cloned and sequenced using standard methods. For details, see Supplementary Text 1.
Quantitative PCR assays
To quantify the MMC, a real-time quantitative PCR assay was developed using the primer pair MMC583f/MMC734r (see above). To relate the abundance of MMC to total bacteria, a 390-bp fragment of the 16S rRNA gene was amplified with the primer pair 517f and 907r (specific for bacterial 16S rRNA genes, Muyzer et al., 1998) following the protocol described by Süß et al. (2006). PCR-generated and purified 16S rRNA gene fragments of a 3.55-kb plasmid containing the 16S rRNA gene of a MMC phylotype obtained from the German Wadden Sea were applied as standards. Abundances of the MMC were determined as percent of total bacterial 16S rRNA genes. Details describing the conditions, technical details and evaluation of the MMC-specific PCR are given in the Supplementary Text 1.
Enrichment and isolation of myxobacteria
As no isolates of the MMC are available, we tried to enrich these organisms in a mesocosm experiment with sediment and water from the Wadden Sea (taken from an intertidal mud flat off the village of Neuharlingersiel on 22 August 2005). To enrich MMC bacteria on artificial surfaces, glass slides were used and prepared as follows: slides were coated with a ca. 1 mm thick layer of agar (1.5%) enriched with peptone (1%). One series of slides was transferred for 6 h in cultures of various bacterial strains (grown in marine broth (Difco, Becton, Dickinson and Company, Franklin Lakes, NJ, USA) up to an OD600 >1) to allow settlement of the cells (as prey organisms). Subsequently, the slides were mounted with a nylon lace to a rod above the mesocosm to keep the slides in the water column about 1 cm above the sediment surface. The slides were incubated in the mesocosm for 3 weeks. Every 2 days, samples were removed from the slides (with a sterile spatula) and DNA was extracted as described above. Presence of bacteria of the MMC in the biofilms was tested with the specific PCR approach.
Additionally, isolation of MMC bacteria from Wadden Sea sediment (taken from an intertidal mud flat off Neuharlingersiel on 30 September 2006) was tried by utilising the bacteriolytic properties of myxobacteria following the methods and using the media described by Reichenbach and Dworkin (1992). Myxobacteria were recognised by formation of fruiting bodies and swarming. Strain MX1 was isolated and purified via several transfers on WCX (water cyxloheximide) agar plates containing 50% of seawater and streaks of dead or living Escherichia coli cells, followed by inoculations on CY (casitone yeast) agar plates, also prepared with 50% of seawater. Strain MX2 was isolated and purified likewise but using WCX agar plates without seawater. Both strains were able to grow on CY agar plates or alternatively on VY/2 (vitamin B12, yeast/2 (half the amount of the normal concentration of VY medium)) agar plates at room temperature and at 30 °C, without and with the addition of 50% of seawater, respectively.
Further details for the enrichment and isolation procedures are given in Supplementary Text 1.
Fosmid library construction and screening for MMC 16S rRNA genes
A fosmid library with DNA from surface sediment of an intertidal sand flat of the German Wadden Sea (see above) was constructed as described by Mussmann et al. (2005) using the EpiFOS fosmid library production kit (Epicenter, Madison, WI, USA) according to the manufacturer's instructions. In total, 11 000 clones from the fosmid library were screened for MMC 16S rRNA genes using the primer pair MMC655f/MMC841r (see above).
Sequencing of the fosmids, ORF finding and sequence annotation
Fosmids were sequenced by a shotgun approach based on plasmid libraries with 1.5–3.5 kb inserts. Sequences were determined by using Big Dye 3.0 chemistry (Applied Biosystems, Foster City, CA, USA), M13 primers (Messing, 1983) and ABI3730XL capillary sequencers (Applied Biosystems) up to a 19-fold coverage. Resulting reads were assembled using the Phrap assembly tool (http://www.phrap.org). All manual editing steps were performed using the GAP4 software package v4.11 (Staden et al., 2000). Prediction of protein-encoding sequences and open reading frames (ORFs) was initially accomplished with YACOP (Tech and Merkl, 2003), producing a combined set of genes predicted by the ORF-finding programs Glimmer (Delcher et al., 1999), Critica (Badger and Olsen, 1999) and Z-curve (Guo et al., 2003). All ORFs were manually curated and verified by comparison with the publicly available databases SwissProt, GenBank, ProDom, COG and Prosite using the annotation software ERGO (Overbeek et al., 2003).
Comparative genomics and bioinformatics tools
The protein sequences encoded by the two fosmids were used for reciprocal BLAST comparisons as well as a global sequence alignment with the Needleman–Wunsch algorithm using the software tool BiBag (pers. comm. Antje Wollherr and Heiko Liesegang, University of Göttingen, Germany). Seven myxobacteria whole-genome protein data sets were taken as query organisms: Anaeromyxobacter dehalogenans strain 2CP-C and strain 2CP-1, Anaeromyxobacter sp. K and sp. Fw109-5, Myxococcus xantus DK1622, S. cellulosum So ce 56 and H. ochraceum DSM14365. Orthologues were identified as reciprocal best BLAST hits with an E-value less than 1e-20, and a Needleman-Wunsch similarity-score more than 25%. Whole sequence alignments and visualisation were performed with the Genome Matcher software (Ohtsubo et al., 2008) using reciprocal BLASTn comparison with a word size of 21 and E-value less than 0.01.
Sequencing of 16S rRNA genes and phylogenetic analysis
PCR products were sequenced using the DYEnamic Direct cycle sequencing kit (Amersham Life Science, Cleveland, OH, USA) and a Model 4200 automated DNA sequencer (LICOR, Lincoln, NE, USA) as described by Rink et al. (2007). Sequences were analysed by BLASTn search (http://www.ncbi.nlm.nih.gov/blast) and the ARB software package (http://www.arb-home.de, Ludwig et al., 2004). A neighbour-joining tree showing the phylogenetic relationships of bacteria of the MMC within the Myxococcales based on 16S rRNA gene sequence similarity was calculated with sequences of at least 1300 bp length (Figure 2). Shorter sequences were added later with maximum parsimony, resulting in Supplementary Figure 1. The origin of all sequences was checked, indicating that they were all retrieved from marine samples. Further details on tree construction and detection of sequences affiliated with the MMC are given in Supplementary Text 1.

Neighbour-joining tree showing the phylogenetic relationships of bacteria of the MMC within the Myxococcales based on 16S rRNA gene sequence similarity. Sequences of at least 1300 bp were considered. Only bootstrap values >50% (derived from 2000 replicates) at main nodes are shown. Filled circles indicate that nodes were also recovered reproducibly with maximum-likelihood. Selected members of the Cyanobacteria were used as outgroup (not shown) to define the root of the tree. GenBank sequence accession numbers are given in parentheses. In addition, the sampling site is indicated. The two clones of the fosmids sequenced in this study and the two new Myxococcus spp. are shown in bold. Bar, 0.10 substitutions per nucleotide position.
The nucleotide sequence data are available at GenBank under accession numbers HQ857564 to HQ857578 (16S rRNA genes), HQ191475 (fosmid MMCf1) and HQ191476 (fosmid MMCf2), GU323922 (Myxococcus sp. MX1) and GU323923 (Myxococcus sp. MX2).
Results
Distribution of the MMC in the North Sea
During a cruise in September 2005 in the North Sea, samples were taken from the water column and the sediment surface. The samples covered a transect from the German Bight to 58°N close to the Norwegian coast (Figure 1a). A specific PCR was used to examine the samples for presence of the MMC. In the water column down to a depth of 3 m above the sediment surface, the MMC was detected only at two stations, once at station 4 (at 15 m depth) and at station 8 where the MMC was present in the whole-water column (see below). In water samples taken during previous cruises in the North Sea (2002 and 2003), the MMC was also detected but only sporadically (Supplementary Table 1). The MMC was detected in all sediment samples taken in 2005 (0 to 0.5 cm horizon), irrespective of the absolute water depth (27–260 m, Supplementary Table 1). Further, the MMC was detected in water samples collected not more than 3 m above the sediment surface at stations 3, 5, 6 and 7. At station 8 with a depth of 29 m, close to the island of Helgoland, the MMC was consistently detected in a time series analysis of a tidal cycle in the particle fraction >5 μm from 5 m depth to the bottom. These results show that the MMC is present in sediments throughout the North Sea and also on resuspended particles in the nepheloid layer.
Abundance of the MMC in sediment samples taken in September 2005 was quantified by real-time quantitative PCR, yielding proportions of 0.8–13.1% of bacterial 16S rRNA genes with highest values at stations 7 and 9–12 (Figure 1b). These results demonstrate that the MMC cluster is not only widely distributed but also a prominent component of the sediment-associated bacterial communities in the North Sea. A coherence of the abundance of MMC bacteria in samples taken on the North Sea transect in 2005 was found with the sediment type, that is, highest numbers of MMC bacteria were found in fine grained sand, compared with medium grained sand to gravel and fine grained sand to muddy sediment (Supplementary Figure 2).
Three cores of sandy sediments in the German Wadden Sea (one core from the Gröninger Plate and two from the Neuharlingersieler Nacken, back-barrier area of Spiekeroog Island, southern North Sea) were examined for presence of the MMC (Supplementary Table 1). It was detected in all three cores to depths of ca. 2 m, indicating its presence not only in oxic but also in anoxic environments, as the sandy sediments in the Wadden Sea become readily anoxic below the surface (Wilms et al., 2006). Analysis of the abundance of the MMC in cores from the Gröninger Plate and the Neuharlingersieler Nacken by quantitative PCR revealed that this cluster constituted 2.8 and 2.0% at the sediment surface, and 4.2 and 1.6% of bacterial 16S rRNA genes in the undermost horizons where the MMC was detected (ca. 2 m sediment depth; for details see Supplementary Table 1). These values are in the same range as those obtained for most sediment surface samples (Figure 1b).
Global distribution and phylogeny of the MMC
To assess the global distribution of the MMC, an analysis of a large set of predominantly sediment samples (n=72, see Supplementary Table 1) was performed. The MMC was detected in almost all samples from marine sediments covering subarctic (Spitzbergen/Svalbard) and temperate regions (eastern and western Atlantic coast, North Sea), the Mediterranean Sea (surface to 4300 m depth), subtropical (Baja California, Florida, Canary Islands, Red Sea, South African coast, Australia) and tropical regions (Caribbean Sea, Ceylon). Only in a few samples of sandy sediments or on macroalgae from various locations (Dominican Republic, Tarquinia (Italy), Red Sea, Majorca, Canary Islands, Khao Lak (Thailand)), at a hydrothermal vent in the Guaymas Basin, as well as in various freshwater habitats the MMC was not detected (Supplementary Table 1).
In total, 11 samples from sediments and cyanobacterial mats from different locations and salinities from 1–164 were also examined for presence of the MMC (Table 1). The results show that its presence is restricted to salinities from 6 to 60 (P<0.05, Chi square χ2-test with Yates correction), emphasising the occurrence of the MMC in marine and brackish but not in freshwater and hypersaline environments.
Phylogenetic analysis of MMC sequences obtained from our samples and closely related sequences found in public databases revealed a distinct cluster within the Myxococcales of exclusively marine, uncultured organisms. Described species in the three myxobacterial suborders are only distantly related to this cluster. Overall, we collected 50 MMC sequences with a length of >1300 bp (Figure 2) and additionally 134 sequences with a length of >450 bp (Supplementary Figure 1). The MMC includes the cluster C13 recently described by Jiang et al. (2010); however, the MMC encompasses more and deeper branching sequences. Robustness of the MMC was demonstrated by a bootstrap value of 99% and the same branching point obtained with a maximum likelihood tree. Sequences obtained with the MMC-specific PCR showed affiliation with the MMC, thus proving specificity of the primers. The deep branching sequence of clone H2 obtained with the specific PCR from North Sea sediment was considered to belong to the MMC, because it was affiliated to clone WHB21–22 (accession number AB426359, Supplementary Figure 1) within the MMC when a maximum likelihood tree was calculated.
Sequences affiliated with the MMC were detected worldwide by us and other authors, for example, in the Arctic Ocean, Greenland, the temperate Pacific coast of the United States of America and Japan, the mid-Atlantic ridge, Panama, Tasmania and Antarctica (Figure 3 and Supplementary Figure 1). Even though most sequences affiliated with the MMC were obtained from sediment samples, these bacteria inhabit also other surfaces, that is, corals, microbial mats and marine benthos.

Map showing the world-wide distribution of the MMC. ▴, detection by the MMC specific PCR, •, detection by sequencing and phylogenetic analysis of 16S rRNA genes obtained in this study and by other authors. The map was adopted from http://www.smithlifescience.com and subsequently modified.
Quantification of the MMC in some of the samples collected worldwide indicated a lower abundance compared with that found in North Sea samples. In a sediment sample taken from the Baltic Sea (sample number 120, Supplementary Table 1), the MMC accounted for 0.71% of the total bacterial community. In other samples, that is, sediment from the eastern Mediterranean Sea (sample no. 167), sediment taken off the coast of North Carolina (sample no. 140) and from a cyanobacterial mat in Baja California (sample no. 186), abundances of the MMC were even lower (0.05%, 0.13% and 0.01%, respectively). This suggests that bacteria of the MMC prosper exceptionally well in North Sea habitats and/or generally in shallow marine sediments rich in organic material, such as intertidal and sublittoral mud flats.
One subcluster of the MMC contains 10 sequences >1300 bp, exclusively obtained from tropical and subtropical locations (Figure 2, subcluster I). Some sequences <1300 bp from other climatic regions, however, also affiliate to this cluster (Supplementary Figure 1, subcluster I). Most other clusters contain a mix of sequences from temperate and polar regions, but seem not to reflect adaptation of subgroups to specific marine environments or conditions.
Within the MMC, organisms appear to be strictly marine. In contrast, sequences between the MMC and the primarily terrestrial described suborders are from marine as well as from other, very diverse habitats, for example, soils, pack ice, a glacier, wastewater treatment plants, compost, a cave wall biofilm and an acid mine drainage (Figure 2).
Enrichment and isolation of myxobacteria
We were able to isolate two strains, Myxococcus sp. MX1 and Myxococcus sp. MX2, from Wadden Sea sediment taken on 30 September 2006. The new strains are affiliated to the Cystobacterineae (Figure 2), with Myxococcus xanthus strain ATCC 25232 as the closest described relative (16S rRNA gene similarity of 98% and 99% to strain MX1 and MX2, respectively) and show also close affiliation to Myxococcus fulvus ATCC 25199, the type species of the genus (16S rRNA gene similarity of 97% and 98% to strain MX1 and MX2, respectively). Our isolation effort with classical isolation techniques for myxobacteria, however, did not yield any organisms of the MMC.
We tried to enrich bacteria of the MMC in a mesocosm experiment with sediment and water from the Wadden Sea. The results indicated presence of bacteria of the MMC on artificial surfaces of agar-coated slides. After 2 days and until the end of the experiment, the MMC-specific PCR gave positive results for all samples removed from the slides, irrespective whether they were presettled with cultivated bacteria (as prey organisms) or not. Formation of biofilms on the agar-coated slides was visible by eye, and strong degradation of the agar became obvious after 2 weeks. Bacteria of the MMC, however, showed no growth after transfer of cells from the slides on agar plates with various media (that is, complex and minimal media with different concentrations of autoclaved seawater and natural sediment, dead marine bacterial strains as prey organisms, cycloheximide to restrict development of fungi, incubation at in situ temperature in the dark).
Sequencing of fosmids, annotation and sequence comparison
Screening of a fosmid library constructed from a sediment sample of the German Wadden Sea revealed two fosmids containing 16S rRNA genes affiliated with the MMC (clone MMCf1 and clone MMCf2, Figure 2). The full sequences of both fosmids were determined. For the 37.1 kb-insert of fosmid MMCf1, 24 protein-encoding ORFs were predicted. The deduced amino-acid sequences of 20 ORFs showed significant similarities (E-value <1e-20) to proteins in the UniProtKB/TrEMBL database (Table 2). The second fosmid, MMCf2, contained an insert of 40.7 kb with 31 predicted protein-encoding ORFs, of which 23 showed significant similarities to proteins in the UniProtKB/TrEMBL database (Table 2). The sequences of both fosmids showed an overlap of ca. 32 kb (Figure 4). In the overlapping region, both fosmids highly resemble each other (89–94% DNA sequence identity). The differences indicate that both fosmids resulted from different strains. The G+C content is 62.3% for MMCf1 and 62.2% for MMCf2. Both fosmids possess a single 16S rRNA gene and no complete rRNA operon as found in most bacteria. Furthermore, no genes coding for transfer RNAs were found besides the 16S rRNA genes as usually found in the internal transcribed spacer regions between 16S and 23S rRNA genes.

Alignment of fosmids MMCf1 and MMCf2. Protein coding ORFs are depicted in light-grey, the 16S rRNA genes in dark-grey. Alignment of the two fosmids is generated by the GenomeMatcher software. The identity score of BLASTn of each block is given in grey scale; white: no similarity; black: highest similarity.
Bidirectional BLAST comparisons of protein sequences of both fosmids with each other and against seven myxobacteria whole-genome protein datasets were carried out. The two fosmids share 20 orthologues (Supplementary Figure 3), 16 of these genes show a similarity score over 90% in the Needleman–Wunsch alignment. For MMCf1, the highest number of orthologues with 14 out of 23 (61%) was identified in two organisms, S. cellulosum and H. ochraceum. Moreover, the ORFs MMCf1_21–22 had the same synteny in the two organisms. This was also observed for MMCf1_23–24 in H. ochraceum and for the two subunits of the highly conserved glutamate synthase (gltAB) in all seven datasets and many genomes of other bacteria from various taxa. A comparison of the genomic loci flanking the glutamate synthase genes revealed no specific genomic locus or any evidence for syntenic gene organisation in other myxobacteria. Besides highly similar ORFs coding for a protein kinase (MMCf1_9) the following ORFs were found in all seven species (Supplementary Figure 3): a nucleoside triphosphatase (MMCf1_12), a conserved hypothetical protein, YyaL-like (MMCf1_13), a putative aldo/keto reductase (MMCf2_9). Nevertheless, the gene order of the two fosmids remains unique among the myxobacteria. The highest number of orthologues for MMCf2 with 15 out of 30 (50%) was assigned to S. cellulosum, closely followed by H. ochraceum with 14 (47%) orthologous genes. Ten ORFs (MMCf1_3/10/17 and MMCf2_3/5/7/10/13/20/27) have no detectable orthologues in any of the references. Interestingly, for nine of these genes (all except MMCf2_10) even no significant BLAST hit in the public databases could be found (Table 2), implying that they are unique for the two fosmids.
The ORFs MMCf1_13 and MMCf2_23 are highly similar to the Desulfobacterium autotrophicum ‘hypothetical protein HRM2_15540’. Orthologues are also present in all completed myxobacteria genomes (Supplementary Figure 3), as well as in other bacteria, archaea and eukaryota and share several domains, for example, a domain of unknown function DUF255 (Pfam03190), a thioredoxin fold (IPR012335) and a six-hairpin glycoside transferase domain (IPR012341). The orthologues of eukaryotic origin are annotated as ‘spermatogenesis-associated proteins’ (for example, UniProtKB/Swiss-Prot entry Q80YT5) for which, according to the Gene Ontology project (GO:0007275, Ashburner et al., 2000), the molecular function is described as ‘multicellular organismal development’, compatible with the myxobacterial life cycle.
In general, genes on the MMCf1 and MMCf2 fosmids could not be assigned to one certain pathway, but seem to be involved in various metabolic routes. Some of the genes (MMCf1_2 and MMCf2_12) might be involved in methylamine metabolism (mauG) or the synthesis of polyamines. The ORFs MMCf1_23 and MMCf1_24 encode a saccharopine dehydrogenase and a carboxynorspermidine decarboxylase. This clustered pair of genes is also present in other bacterial lineages and most probably part of an alternative route for the synthesis of both sym-norspermidine and spermidine (Lee et al., 2009). Other genes (MMCf1_6/7 and MMCf2_16/17) are likely part of amino-acid metabolism (glutamate synthase, gltAB).
Discussion
Similar to Gram-positive bacteria, myxobacteria have been considered for a long time as typical terrestrial organisms. Strains of both groups sporadically isolated from marine habitats were thought not to be indigenous but introduced from terrestrial habitats (Zobell and Upham, 1944; Goodfellow and Haynes, 1984; Reichenbach, 1999; Dawid, 2000). Today, it is widely accepted that distinct marine clusters of Gram-positive bacteria exist, which are only distantly related to clusters comprising also Gram-positive bacteria from freshwater and soil (for example, Rappé et al., 1999; Mincer et al., 2002). Here, we demonstrated that bacteria of the MMC are distributed worldwide in marine sediments and also present on several other marine surfaces but are absent in non-marine habitats. Thus, our study confirms the existence of distinct marine myxobacterial clusters (Jiang et al., 2010) and shows that they evolved separately from the known myxobacterial suborders. Furthermore, it extends our knowledge about the biogeography and diversity of these organisms and allowed a first insight into their genomic features. Phylogenetic analysis revealed that sequences that classify the phylogenetic tree between the MMC and the primarily terrestrial clusters of the known suborders were obtained from marine but also from other, very diverse non-saline habitats (Figure 2). This overlap might reflect a transition and adaptation from a terrestrial to a marine lifestyle.
The MMC is only sporadically present in the water column, and if so, then mostly on aggregates close to the sediment surface. Thus resuspension of sediment particles by tidal currents (Jago et al., 2002) is a possible reason for presence of these organisms in the water column. The MMC was predominantly found in oxic habitats, suggesting an aerobic metabolism as described for most myxobacteria (Shimkets et al., 2006). On the basis of the presence of MMC bacteria in three different sediment cores from the North Sea down to a sediment depth of about 2 m, however, one can speculate that these organisms can also live under anoxic conditions or produced spores that were buried in the sediment. Oxygen is only present in the upper 3 mm of the sediment at the sampling sites (Köpke et al., 2005). Myxobacteria have been known for a long time to produce spores (Shimkets, 1990; Shimkets et al., 2006), but in contrast to other organisms of the Deltaproteobacteria, most myxobacteria are strictly aerobic heterotrophs. A. dehalogenans (Cystobacterineae), however, was shown to be facultatively anaerobic, using acetate, H2, succinate, pyruvate, formate and lactate as electron donors, and various chlorophenolic compounds and nitrate as electron acceptors (Sanford et al., 2002). To elucidate how MMC bacteria survive in anoxic sediment, further studies are needed, that is, isolation of these organisms and subsequent physiological characterisation or molecular and gene expression analyses to investigate their metabolic activity.
By applying classical isolation methods, we were able to obtain two isolates affiliated with the Myxococcaceae (within Cystobacterineae) from the Wadden Sea sediment. Myxococcus strains have been obtained from marine environments before and were investigated for their adaptation to marine conditions. Isolates obtained by Wang et al. (2007) possessed different levels of salt tolerance, had the dual motility system and formed fruiting bodies in the presence of suitable seawater concentrations. Some high salt-tolerant strains even lost their fruiting abilities in the absence of seawater Wang et al. (2007). Overall, the results suggested an ecological adaptation of myxobacterial social behaviours of these isolates to marine environments (Wang et al., 2007). With the classical methods used in this study (Reichenbach and Dworkin, 1992), no strains of the MMC were obtained what might explain why no organisms of this cluster have been isolated by other researchers so far. Whether the two new Myxococcus strains MX1 and MX2 are real marine organisms remains open, because currently we only know that they can grow with and without the addition of 50% of seawater to the medium.
In the mesocosm experiment, we were able to induce at least settlement of MMC bacteria on agar-coated slides. In contrast to the classical methods, the mesocosm experiment provided an environment more similar to natural conditions and thus allowed survival or even growth of MMC bacteria. Settlement occurred not only on slides presettled with cultivated bacteria but also on pure agar. As other bacteria certainly also settled on these slides, we can not determine whether MMC bacteria are attracted by the presence of other microorganisms or if they even hydrolyse other bacteria. The experiment, however, confirmed the preference of MMC bacteria to settle on various surfaces.
Sequence analysis of two MMC fosmids found in this study indicated that both are highly similar but arose from different strains. The GC content of the fosmid sequences is only slightly lower than that of known genomes of other myxobacteria (64 to 72 mol %). Both fosmids have a single 16S rRNA gene and no complete rRNA operon. Split rRNA operons have been identified in bacteria and archaea previously, primarily when an entire genome has been sequenced. In the majority of cases, the 23S and 5S genes form an operon and the 16S rRNA gene is separate, and there can be one or more than one copy of each of the genes (Boyer et al., 2001). Even though unlinked rRNA operons are rare, they are known for several organisms affiliated with Planctomycetes, Deinococcus-Thermus, Alpha-, Gamma- and Epsilonproteobacteria, Poribacteria or Euryarchaeota (http://www.ncbi.nih.gov/genomes/lproks.cgi; https://img.jgi.doe.gov/cgi-bin/er/main.cgi; Liesack and Stackebrandt, 1989; Ruepp et al., 2000; Tamas et al., 2002; Glöckner et al., 2003; Wu et al., 2003; Henne et al., 2004; Fieseler et al., 2006). For Deltaproteobacteria, however, this is to our knowledge the first description of an unlinked rRNA operon. Unusual organisation of rRNA operons is known for myxobacteria. The genome of S. cellulosum (account no. AM746676; Schneiker et al., 2007) contains two complete rRNA operons (16S-23S-5S), one incomplete (16S-23S) and one with two 5S rRNA genes (16S-23S-5S-5S).
Comparison of the genomic loci flanking the 16S rRNA genes on the two fosmids MMCf1 and MMCf2 revealed neither a specific genomic locus nor any evidence for a syntenic gene organisation. Although both fosmids have 50% and 61% orthologues in other sequenced myxobacteria, respectively (Supplementary Figure 3), it is striking that about half of the closest homologues of the genes identified on the fosmids by BLASTp analysis were found in other marine bacteria from various taxa and less than one-third of the closest homologues were found within the myxobacteria (Table 2). This might reflect an ancient separation of the MMC from other myxobacterial lineages and adaptation to the marine environment. For nine of the genes identified on the fosmids, no significant BLAST hits in the public databases were found (Table 2). This indicates that at least on a genomic basis bacteria of the MMC are exceptional. Sequencing of the fosmids allowed the identification of other than 16S rRNA genes. Thus, in future studies, highly specific primer systems can be used that target not only the 16S rRNA gene, allowing a higher specificity, as well as expression analyses of functional genes to monitor the activity and to further elucidate details of the lifestyle of these unique bacteria.
Overall, distribution and abundance of the MMC suggest that it comprises important players in marine sediment-associated bacterial communities. Phylogenetic analysis revealed one subcluster (subcluster I, Figure 2) obtained exclusively from tropical and subtropical regions, which might reflect adaptation to specific conditions. Except for a coherence of abundance with the sediment type, and presence with salinity, no further parameters influencing the distribution were identified. Their physiology remains also largely unknown, and we currently do not know whether they hydrolyse biopolymers or even prey on other microorganisms, similar to terrestrial myxobacteria. MMC bacteria also could be a novel source of secondary metabolites because their terrestrial counterparts are one of the prime sources of these bioactive compounds (Gerth et al., 2003; Wenzel and Müller, 2007). Future studies have to reveal this possible activity, the ecological role of the MMC, parameters having influence on its distribution and whether these organisms are a mortality factor for bacterial growth not considered so far in marine sediments.
References
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al. (2000). Gene ontology: tool for the unification of biology. Nat Genet 25: 25–29.
Badger JH, Olsen GJ . (1999). CRITICA: coding region identification tool invoking comparative analysis. Mol Biol Evol 16: 512–524.
Boyer SL, Flechtner VR, Johansen JR . (2001). Is the 16S–23S rRNA internal transcribed spacer region a good tool for use in molecular systematics and population genetics? A case study in cyanobacteria. Mol Biol Evol 18: 1057–1069.
Dawid W . (2000). Biology and global distribution of myxobacteria in soils. FEMS Microbiol Rev 24: 403–427.
Delcher AL, Harmon D, Kasif S, White O, Salzberg SL . (1999). Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27: 4636–4641.
Fieseler L, Quaiser A, Schleper C, Hentschel U . (2006). Analysis of the first genome fragment from the marine sponge-associated, novel candidate phylum Poribacteria by environmental genomics. Environ Microbiol 8: 612–624.
Foudou R, Jojima Y, Yamanaka S . (2002). Haliangium ochraceum gen. nov., sp. nov. and Haliangium tepidum sp. nov.: novel moderately halophilic myxobacteria isolated from coastal saline environments. J Gen Appl Microbiol 48: 109–116.
Garcia R, Gerth K, Stadler M, Dogma IJ, Müller R . (2010). Expanded phylogeny of myxobacteria and evidence for cultivation of the ‘unculturables’. Mol Phylogenet Evol 57: 878–887.
Gerth K, Pradella S, Perlova O, Beyer S, Müller R . (2003). Myxobacteria: proficient producers of novel natural products with various biological activities - past and future biotechnological aspects with the focus on the genus Sorangium. J Biotechnol 106: 233–253.
Giebel HA, Brinkhoff T, Zwisler W, Selje N, Simon M . (2009). Distribution of Roseobacter RCA and SAR11 lineages and distinct bacterial communities from the subtropics to the Southern Ocean. Environ Microbiol 11: 2164–2178.
Glöckner FO, Kube M, Bauer M, Teeling H, Lombardot T, Ludwig W et al. (2003). Complete genome sequence of the marine planctomycete Pirellula sp. strain 1. Proc Natl Acad Sci USA 100: 8298–8303.
Goldman BS, Nierman WC, Kaiser D, Slater SC, Durkin AS, Eisen JA et al. (2006). Evolution of sensory complexity recorded in a myxobacterial genome. Proc Natl Acad Sci 103: 15200–15205.
Goodfellow M, Haynes JA . (1984). Actinomycetes in marine sediments. In Ortiz-Ortiz L (ed), Biological, Biochemical and Biomedical Aspects of Actinomycetes. Academic Press: New York, NY, USA, pp 453–472.
Guo FB, Ou HY, Zhang CT . (2003). ZCURVE: a new system for recognizing protein-coding genes in bacterial and archaeal genomes. Nucleic Acids Res 31: 1780–1789.
Henne A, Bruggemann H, Raasch C, Wiezer A, Hartsch T, Liesegang H et al. (2004). The genome sequence of the extreme thermophile Thermus thermophilus. Nat Biotechnol 22: 547–553.
Iizuka T, Jojima Y, Fudou R, Hiraishi A, Ahn JW, Yamanaka S . (2003a). Plesiocystis pacifica gen. nov., sp. nov., a marine myxobacterium that contains dihydrogenated menaquinone, isolated from the Pacific coasts of Japan. J System Evol Microbiol 53: 189–195.
Iizuka T, Jojima Y, Fudou R, Tokura M, Hiraishi A, Yamanaka S . (2003b). Enhygromyxa salina gen. nov., sp. nov., a slightly halophilic Myxobacterium isolated from the coastal areas of Japan. System Appl Microbiol 26: 189–196.
Jago CF, Jones SE, Latter RJ, McCAndliss RR, Hearn MR, Howarth MJ . (2002). Resuspension of benthic fluff by tidal currents in deep stratified waters, northern North Sea. J Sea Res 48: 259–269.
Jiang DM, Kato C, Zhou XW, Wu ZH, Sato T, Li YZ . (2010). Phylogeographic separation of marine and soil myxobacteria at high levels of classification. ISME J 4: 1520–1530.
Köpke B, Wilms R, Engelen B, Cypionka H, Sass H . (2005). Microbial diversity in coastal subsurface sediments: a cultivation approach using various electron acceptors and substrate gradients. Appl Environ Microbiol 71: 7819–7830.
Lee J, Sperandio V, Frantz DE, Longgood J, Camilli A, Phillips MA et al. (2009). An alternative polyamine biosynthetic pathway is widespread in bacteria and essential for biofilm formation in Vibrio cholerae. J Biol Chem 284: 9899–9907.
Liesack W, Stackebrandt E . (1989). Evidence for unlinked rrn operons in the planctomycete Pirellula marina. J Bacteriol 171: 5025–5030.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
Messing J . (1983). New M13 vectors for cloning. Method Enzymol 101: 20–78.
Mincer TJ, Jensen PR, Kauffman CA, Fenical W . (2002). Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments. Appl Environ Microbiol 68: 5005–5011.
Mussmann M, Richter M, Lombardot T, Meyerdierks A, Kuever J, Kube M et al. (2005). Clustered genes related to sulfate respiration in uncultured prokaryotes support the theory of their concomitant horizontal transfer. J Bacteriol 187: 7126–7137.
Muyzer G, Brinkhoff T, Nübel U, Santegoeds C, Schäfer H, Wawer C . (1998). Denaturing gradient gel electrophoresis (DGGE) in microbial ecology. In Akkermans ADL, van Elsas JD, de Brujin FJ (eds). Molecular Microbial Ecology Manual. 3rd edn Kluwer Academic Publishers: Dordrecht, The Netherlands, pp 1–27, p. 3.4.4.
Muyzer G, Teske A, Wirsen CO, Jannasch HW . (1995). Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis. Arch Microbiol 164: 165–172.
Ohtsubo Y, Ikeda-Ohtsubo W, Nagata Y, Tsuda M . (2008). GenomeMatcher: a graphical user interface for DNA sequence comparison. BMC Bioinformatics 9: 376.
Overbeek R, Larsen N, Walunas T, D’Souza M, Pusch G, Selkov E et al. (2003). The ERGO genome analysis and discovery system. Nucleic Acids Res 31: 164–171.
Rappé MS, Gordon DA, Vergin KL, Giovannoni SJ . (1999). Phylogeny of actinobacteria small subunit (SSU) rRNA gene clones recovered from marine bacterioplankton. Syst Appl Microbiol 22: 106–112.
Reichenbach H . (1999). The ecology of the myxobacteria. Environ Microbiol 1: 15–21.
Reichenbach H . (2005). Bergey's Manual of Systematic Bacteriology 2nd edn, 2: 1059–1144.
Reichenbach H, Dworkin M . (1992). The myxobacteria. In Balows A et al. (eds). The Prokaryotes. 2nd edn, Vol 4. Springer Verlag: New York, pp 3416–3487.
Rink B, Seeberger S, Martens T, Duerselen CD, Simon M, Brinkhoff T . (2007). Effects of a phytoplankton bloom in a coastal ecosystem on the composition of bacterial communities. Aquat Microb Ecol 48: 47–60.
Rückert G . (1984). Untersuchungen zum Vorkommen von Myxobakterien in von Meerwasser beeinflußten Substraten unter besonderer Berücksichtigung der Insel Helgoland. Helgoländer Meeresunters 38: 179–184.
Ruepp A, Graml W, Santos-Martinez ML, Koretke KK, Volker C, Mewes H et al. (2000). The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature 407: 508–513.
Sanford RA, Cole JR, Tiedje JM . (2002). Characterization and description of Anaeromyxobacter dehalogenans gen. nov., sp. nov., an aryl-halorespiring facultative anaerobic myxobacterium. Appl Environ Microbiol 68: 893–900.
Schneiker S, Perlova O, Kaiser O, Gerth K, Alici A, Altmeyer MO et al. (2007). Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat Biotechnol 25: 1281–1289.
Shimkets LJ . (1990). Social and developmental biology of the myxobacteria. Microbiol Rev 54: 473–501.
Shimkets LJ, Dworkin M, Reichenbach H . (2006). The myxobacteria. In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds). The Prokaryotes. 3rd edn vol. 7. Springer: Heidelberg, pp 31–115.
Staden R, Beal KF, Bonfield JK . (2000). The Staden package, 1998. Methods Mol Biol 132: 115–130.
Stevens H, Brinkhoff T, Simon M . (2005). Composition and seasonal dynamics of free-living, aggregate- and sediment surface-associated bacterial communities in the German Wadden Sea. Aquat Microb Ecol 38: 15–30.
Süß J, Schubert K, Sass H, Cypionka H, Overmann J, Engelen B . (2006). Widespread distribution and high abundance of Rhizobium radiobacter within Mediterranean subsurface sediments. Environ Microbiol 8: 1753–1763.
Tamas I, Klasson L, Canback B, Naslund AK, Eriksson AS, Wernegreen JJ et al. (2002). 50 million years of genomic stasis in endosymbiotic bacteria. Science 296: 2376–2379.
Tech M, Merkl R . (2003). YACOP: enhanced gene prediction obtained by a combination of existing methods. In Silico Biol 3: 441–451.
Wang B, Hu W, Liu H, Zhang CY, Zhao JY, Jiang DM et al. (2007). Adaptation of salt-tolerant Myxococcus strains and their motility systems to the ocean conditions. Microb Ecol 54: 43–51.
Weissman KJ, Müller R . (2010). Myxobacterial secondary metabolites: bioactivities and modes-of-action. Nat Prod Rep 27: 1276–1295.
Wenzel SC, Müller R . (2007). Myxobacterial natural product assembly lines: fascinating examples of curious biochemistry. Nat Prod Rep 24: 1211–1224.
Wilms R, Köpke B, Sass H, Chang TS, Cypionka H, Engelen B . (2006). Deep biosphere-related bacteria within the subsurface of tidal flat sediments. Environ Microbiol 8: 709–719.
Wu M, Sun L, Vamathevan J, Riegler M, DeBoy RT, Brownlie J et al. (2003). Direct submission to the NCBI database (www document) http://www.ncbi.nlm.nih.gov/genomes/framik.cgi?db=Genome&gi=383.
Zhang YQ, Li YZ, Wang B, Wu ZH, Zhang CY, Gong X et al. (2005). Characteristics and living patterns of marine myxobacterial isolates. Appl Environ Microbiol 71: 3331–3336.
Zhou J, Bruns MA, Tiedje JM . (1996). DNA recovery from soils of diverse composition. Appl Environ Microbiol 62: 316–322.
Zobell CE, Upham HC . (1944). A list of marine bacteria including descriptions of sixty new species. Bull Scripps Inst Oceanogr Univ Calif 5: 239–292.
Acknowledgements
We thank the crew of the RV Heincke for their valuable support on shipboard, Thomas Badewien and Axel Braun for technical assistance in sampling and Helge Giebel for help with graphical presentations. Furthermore, we thank numerous colleagues and friends for providing samples from various locations. This work was supported by a grant from the Deutsche Forschungsgemeinschaft within the Research Unit ‘BioGeoChemistry of the Wadden Sea’ (FG-432 TP-5 and TP-B).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Brinkhoff, T., Fischer, D., Vollmers, J. et al. Biogeography and phylogenetic diversity of a cluster of exclusively marine myxobacteria. ISME J 6, 1260–1272 (2012). https://doi.org/10.1038/ismej.2011.190
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2011.190
Keywords
This article is cited by
-
Diversity of Myxobacteria Isolated from Indonesian Mangroves and Their Potential for New Antimicrobial Sources
Current Microbiology (2023)
-
Analysis of the Genome and Metabolome of Marine Myxobacteria Reveals High Potential for Biosynthesis of Novel Specialized Metabolites
Scientific Reports (2018)
-
Current trends in myxobacteria research
Annals of Microbiology (2016)
-
Metagenomic analysis of size-fractionated picoplankton in a marine oxygen minimum zone
The ISME Journal (2014)
