
Abstract
Prokaryotic communities inhabiting surface waters of temperate areas exhibit patterns of seasonal succession. Generally, studies describing these temporal changes are not performed in the proximity to the coast. In the present study, temporal variation of these communities was determined in surface waters at two stations located in the close proximity to the eastern shore of the northern Adriatic Sea. Sequencing of the V4 region of the 16S rRNA gene identified the highest community richness in December with distinct shifts in community structure between periods from April to May, June to October, and November to March. Temperature was shown to be the main environmental force explaining community temporal variation. The NS5 marine group, uncultured Cryomorphaceae, SAR86 clade, and Synechococcus were present throughout the year. Members without know relatives within Rhodobacteraceae and the NS4 marine group were more pronounced in the period from April to May, the AEGEAN-169 marine group, SAR11 subclade III, and HIMB11 in the period from June to October, and SAR11 subclade Ia and Archaea in the period from November to March. Litoricola and OM60 (NOR5) clade were characteristic for both the community sampled from April to May and November to March. Taken together, prokaryotic communities inhabiting nearshore surface waters exhibit a general pattern in community structure similar to other surface associated assemblages of temperate areas. However, the identified specific community composition and temporal patterns differ from other coastal areas.
Similar content being viewed by others

Introduction
Prokaryotic picoplankton communities inhabiting marine surface waters exhibit seasonal succession. These temporal community changes were described for surface waters of polar, temperate, and (sub)tropical regions1. In temperate regions changes were mainly associated with summer water column stratification, winter mixing, and spring phytoplankton blooms1,2,3. Although general successional patterns in these waters have been reported, some local differences were also observed. While some studies have reported the exchange of multiple community states during the year4,5,6,7, others have observed a community separation in only two major groups, specifically the period from November to April (colder months) and from May to October (warmer months)2. Beside global patterns local conditions such as the presence and intensity of phytoplankton blooms3,8,9 or terrestrial nutrient inputs10 may influence seasonal community change.
Seasonal community sucession in temperate waters usually starts with assemblages characteristic for spring phytoplankton blooms. The successional pattern of different microbial groups during the pre-bloom, bloom, and bloom-decay periods have been described in detail3,5,9. The pre-bloom community is generally dominated by members of the alphaproteobacterial SAR11 clade, during the bloom taxa within Bacteroidota, such as Formosa, Polaribacter, Ulvibacter, and the VIS6 clade, become abundant, while the decay period is characterised by Gammaproteobacteria, i.e. the SAR92 clade3,5,9. Beside taxa co-occurring with phytoplankton blooms, communities specific to summer water stratification, characterised by the higher presence of Flavobacteria and Synechococcus, and communities specific for winter mixing, characterised by the higher relative abundance of the SAR11 clade, were described2. In addition, it was found that some subclades of SAR11 such as the subclade Ia are characteristic for summer and some such as subclades Ib and II for winter months11.
The majority of studies describing temporal changes in temperate areas were performed at long-term time series stations, such as the L4 sampling site of the Western Channel Observatory located in the Western English Channel4,12, Blanes Bay Microbial Observatory (BBMO) located in the northwestern Mediterranean2,13, Linnaeus Microbial Observatory located in the Baltic Sea6, station Kabeltonne in the German Bight (southeastern North Sea)3,9, and station E2 of the RADIALES time-series project located in the southern part of the Bay of Biscay14. All these stations, with the exception of station Kabeltonne, are located 0.5 nautical miles or more from the shore. In addition, seawater samples used in these studies were retrieved from surface waters. Data obtained from these time-series studies have found that a set of abiotic and biotic factors drive the temporal community variation1. It was suggested that biological interactions primarily affect microbial dynamics over time periods of days to weeks, while physicochemical parameters, such as light, temperature, and inorganic nutrient concentration, are mainly responsible for observed seasonal successional patterns1,2,4,12,15,16. In addition, several studies performed in surface waters of: station Kabeltonne, the San Pedro Ocean Time series location, and the Linnaeus Microbial Observatory indicate that phytoplankton derived dissolved organic matter (DOM) drives community dynamics1,3,6,9,15. It is therefore worth investigating whether such general interactions also apply to nearshore microbial communities in other areas such as the northern Adriatic Sea.
Surface waters at two stations along the eastern coast of the northern Adriatic Sea were sampled at monthly intervals to determine the temporal variation of prokaryotic picoplankton communities in these habitats. In addition, to assess the main environmental parameters associated with community change, compositional data were linked to a set of previously reported environmental parameters measured at the same time17,18.
Results
Sequencing of 17 samples from the Bay of Saline and 18 samples from the Bay of Funtana (one of the samples was a sequencing replicate) yielded 1.5 million reads after quality curation and exclusion of sequences without known relatives (no relative sequences), eukaryotic, chloroplast, and mitochondrial sequences (Supplementary Table S1). The number of reads per sample ranged from 25,360 to 77,466 (Supplementary Fig. S1 and Table S1). Reads were clustered into 16,629 different OTUs at a similarity level of 97 %. To account for different sequencing depth reads were normalized to the minimum number of sequences per sample (25,360, Supplementary Table S1) that resulted in 13,440 different OTUs ranging from 608 to 1790 OTUs per sample (Supplementary Fig. S2).
Temporal variations in richness and diversity were determined by calculating the observed number of OTUs, Chao1 and ACE richness estimators, and Exponential Shannon and Inverse Simpson diversity indices. Similar trends in richness and diversity were observed at both stations (Supplementary Fig. S2) characterised by a maximum richness in both, the Saline (Observed Number of OTUs, 1790 OTUs) and Funtana (Observed Number of OTUs, 1786 OTUs) Bay in December 2017. In contrast, the Inverse Simpson index did not show an elevated value in December 2017 indicating that rare OTUs contributed substantially to the observed richness maxima. To determine temporal changes in the proportion of shared OTUs and communities the Jaccard’s and Bray-Curtis similarity coefficients were calculated between consecutive sampling points (Supplementary Fig. S3). Similar trends were observed at both stations with higher stability of shared bacterial and archaeal OTUs (Jaccard’s similarity coefficient) than shared communities (Bray-Curtis similarity coefficient). A substantial decline in community similarity between March and April 2018 was observed at both stations indicating a pronounced community shift in this period (Supplementary Fig. S3). Analysis of this time series data showed that only 0.6 % of OTUs were present throughout the study period, while these persistent OTUs contributed to 62.0 % of sequences. Taxonomic classification of these reads revealed that they mainly contributed to abundant phylogenetic groups described below (Supplementary Table S2).
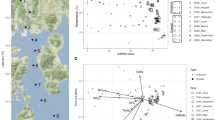
To evaluate the temporal variation of bacterial and archaeal communities Principal Coordinate Analysis (PCoA) was computed using Bray-Curtis dissimilarities based on OTU abundances (Fig. 1a). We identified regardless of the station sampled three separate communities: one specific for the period from June to October, one for the period from November to March, and one for the period from April to May. This separation into three specific communities was further supported by ANOSIM (R = 0.95, P < 0.01). To assess which environmental parameter mainly contributes to the observed temporal community variation, the community data were constrained by a set of environmental variables using distance-based Redundancy Analysis (db-RDA) (Fig. 1b). Nearly half (\(R^2_a=\) 45.6 %) of the observed community variation could be explained by all the variables. Temperature, prokaryotic abundance, salinity, and nitrite mainly explained the separation between communities except for the community encompassing the period from April to May, whose separation could not be explained by any variable.

(a) Principal Coordinates Analysis (PCoA) of Bray-Curtis dissimilarities based on OTU abundances of bacterial and archaeal communities sampled in the Bay of Saline and Funtana. The proportion of explained variation by each axis is shown on the corresponding axis in parentheses. (b) Distance-based Redundancy Analysis (db-RDA) of Bray-Curtis dissimilarities based on the same community data sampled at the same locations and constrained by a set of environmental parameters (T – temperature, S – salinity, \(\hbox {PO}_{4}^{3-}\) – orthophosphate, \(\hbox {NH}_{4}^{+}\) – ammonium, \(\hbox {NO}_{2}^{-}\) – nitrite, \(\hbox {NO}_{3}^{-}\) – nitrate, \(\hbox {Si(OH)}_{4}\) – silicic acid, PM – particulate matter, Chl a – chlorophyll a, and PA – prokaryotic abundance). Scaling type 2 and fitted site scores were selected to construct the plot. The proportion of community data variation explained by environmental variables (\(R^2_a\)) is stated on the biplot, while the proportion of community data variation explained by each canonical axis is shown on the corresponding axis in parenthesis. Samples in both plots originating from different months, years, and stations are labeled in different shape and color.

Taxonomic classification and relative contribution of the most abundant (≥ 1 %) bacterial and archaeal sequences during different time periods. No Relative – sequences without known relatives.
The classification of reads showed that the prokaryotic community was dominated by bacterial (97.8 ± 3.8 %) over archaeal sequences (1.8 ± 3.5 %) (Fig. 2). The bacterial community was comprised of well-known seawater groups such as the Actinobacteriota, Bacteroidota, Cyanobacteria, Marinimicrobia, Alphaproteobacteria, Gammaproteobacteria, and Verrucomicrobiota (Fig. 2). Sequences classified as Alphaproteobacteria showed the highest relative abundance and comprised on average 38.3 ± 8.0 % of the prokaryotic community. The relative contribution of Alphaproteobacteria was higher in the period from June to October (40.2 ± 6.5 %) and November to March (39.7 ± 5.6 %) than in the period from April to May (25.5 ± 9.5 %) (Figs. 2 and 3a). Taxa within this group showed substantial variation between different temporal communities. The alphaproteobacterial community in the period from April to May was characterised by Ascidiaceihabitans (18.0 ± 6.8 %), no relative Rhodobacteraceae (17.2 ± 13.8 %), and Stappiaceae (11.7 ± 12.6 %), in the period from June to October by the AEGEAN-169 marine group (21.4 ± 7.7 %), SAR11 subclade III (15.9 ± 8.9 %), and HIMB11 (11.3 ± 5.1 %), while in the period from November to March by SAR11 subclade Ia (42.0 ± 4.7 %) (Fig. 3a).

Taxonomic classification and relative contribution of the most abundant sequences within Alphaproteobacteria (≥ 2 %) (a), Bacteroidota (≥ 1 %) (b), Gammaproteobacteria (\(\ge\) 1 %) (c), and Cyanobacteria (≥ 1 %) (d) during different time periods. The proportion of sequences classified into each of these taxa in the total bacterial and archaeal community is given above the corresponding bar. NR – No Relative (sequences without known relatives within the corresponding group).
Bacteroidota comprised on average 21.8 ± 6.2 % of the community. Higher values were found in the period from June to October (25.0 ± 4.7 %) and April to May (20.8 ± 3.7 %) than in the period from November to March (13.7 ± 2.2 %) (Figs. 2 and 3b). Some taxonomic groups within Bacteroidota, such as the NS5 marine group (30.7 ± 10.3 – 38.9 ± 11.4 %) and uncultured Cryomorphaceae (11.1 ± 5.1 – 14.6 ± 6.4 %), were characteristic for all the identified temporal communities, while others showed higher presence in one of the communities. For example, the NS4 marine group (30.9 ± 13.8 %) showed the highest contribution to the Bacteroidota community in the period from April to May, while uncultured Flavobacteriaceae (8.3 ± 6.3 %), Balneola (6.5 ± 3.8 %), the NS11-12 marine group (5.4 ± 1.7 %), and uncultured Balneolaceae (4.8 ± 4.0 %) were more pronounced in the period from June to October (Fig. 3b).
Reads classified as Gammaproteobacteria comprised on average 21.6 ± 6.6 % of the prokaryotic community. In contrast to Alphaproteobacteria, gammaproteobacterial sequences showed a higher relative abundance in the period from April to May (35.7 ± 11.1 %) than in other periods (June – October, 19.6 ± 2.5 % and November – March, 20.3 ± 2.8 %) (Figs. 2 and 3c). Within Gammaproteobacteria SAR86 clade was present throughout the study period (20.7 ± 10.5 – 34.8 ± 2.5 %). Litoricola and OM60 (NOR5) clade characterised the period from April to May (Litoricola, 32.4 ± 15.4 % and OM60 (NOR5) clade, 17.4 ± 2.3 %) and June to October (Litoricola, 12.3 ± 8.5 % and OM60 (NOR5) clade, 19.3 ± 5.1 %), while the SUP05 cluster was specific to the period from November to March (7.9 ± 4.2 %) (Fig. 3c).
Cyanobacteria comprised on average 5.1 ± 2.8 % of the total community. No large differences were found in the proportion of cyanobacterial sequences between the analysed periods (4.0 ± 2.4 – 5.4 ± 1.6 %) (Figs. 2 and 3d). Throughout the study period the cyanobacterial community was largely dominated by Synechococcus (82.7 ± 15.6 – 96.1 ± 3.0 %) (Fig. 3d). A higher relative contribution of archaeal reads was observed only in the period from November to March (6.4 ± 5.0 %), while in other periods their proportion in the total community was low (April – May, 0.2 ± 0.2 % and June – October, 0.4 ± 0.2 % (Fig. 2). The main taxonomic groups within Archaea contributing to the higher relative abundance of archaeal sequences in the period from November to March were the Thermoplasmatota Marine group II (56.2 ± 15.2 %) and Crenarchaeota “Candidatus Nitrosopumilus” (31.4 ± 20.8 %).
Discussion
Prokaryotic communities inhabiting surface waters of polar, temperate, and (sub)tropical regions exhibit patterns of seasonal succession1. These temporal variations were mainly studied at long-term time series sites usually encompassing only one sampling station located further away from the coast2,4. In the present study the temporal variation of surface prokaryotic communities was determined at two sites in the close proximity of the shore.
Temporal changes in OTUs were considerable as indicated by the low proportion of OTUs present at each sampling date (0.6 %). This low number of persistent OTUs, however, comprised a high proportion of reads (62.0 %). Similar proportions of persistent core OTUs and their contribution to the total number of sequences were also reported in other time series studies4,12. Interestingly, the most abundant persistent OTUs were classified into taxa showing the highest proportion of reads. Analysis of the temporal variations in alpha-diversity showed maximal richness in December (Supplementary Fig. S2). This observation is in agreement with previously reported richness maxima in other regions during colder months2,7,12,19. It has been suggested that late autumn/winter overturn is responsible for this phenomenon by simply mixing populations from deeper parts of the water column with existing ones1,11,20. However, a similar richness pattern was also observed in regional seas where seasonal overturning of the water column does not play a role, such as in the shallow North Sea where also a higher richness was observed in winter21. Although the samples in this study were retrieved at very shallow locations, water column mixing taking place at deeper areas of the Adriatic Sea sustained with the circulation could bring additional taxa to these locations causing the observed increase in alpha-diversity.
We identified three distinct microbial assemblages characteristic for the period from April to May, June to October, and November to March (Fig. 1a). This is in agreement with studies describing the exchange of multiple communities over an annual cycle with a distinct spring community assemblage6,7 and in contrast to studies describing only a switch between winter- and summer-specific assemblages2,8. The distinct community detected in the period from April to May is likely a response to a phytoplankton bloom that can occur in this region22,23 as it was reported that the absence of spring and fall phytoplankton blooms in some areas can lead to a lower number of microbial assemblages8. Temperature and prokaryotic abundance were identified as main factors influencing the exchange of communities between the period from June to October and the period from November to March (Fig. 1b). It is not surprising that temperature and prokaryotic abundance almost equally explain this shift as higher prokaryotic abundances were reported in this area during summer months24. The identification of temperature as the single most important driver of community change is in line with previously reported data2,7,8. It was proposed that temperature indirectly influences community change through phytoplankton nutrient limitation during water column stratification and nutrient input in times of water column mixing1.
Taxonomic analysis revealed taxa characteristic for all the identified temporal communities, but also taxa characteristic for only one or two of the communities (Figs. 2 and 3). The flavobacterial NS5 marine group and uncultured Cryomorphaceae, the gammaproteobacterial SAR86 clade, and cyanobacterial Synechococcus were characteristic for all the identified communities. Cryomorphaceae are associated with organic matter re-mineralisation processes25, while a single-cell genome analysis of the NS5 marine group revealed its ability to degrade marine polysaccharides26. In addition, the NS5 marine group was previously detected in different seasons and environments of the Adriatic Sea27,28. These two groups could be a part of a basic re-mineralisation community present at this location throughout the year. The gammaproteobacterial SAR86 clade, previously reported in different environments of the Adriatic Sea10,27,28, was also detected in all communities. Recent analysis of metagenomic data suggests the existence of different functional and ecological ecotypes of this ubiquitous clade29. It is possible that different ecotypes are also characteristic for different seasons. The dominance of Synechococcus over other cyanobacterial groups in this coastal area was reported previously10,30. The known genome versatility of Synechococcus could explain the high contribution of this genus to the cyanobacterial community in fluctuating coastal environments31.
Differences between communities specific for the period from April to May, June to October, and November to March observed at the level of OTUs could also be seen in the taxonomic composition (Figs. 2 and 3). The identified community from April to May was characterised by the alphaproteobacterial Ascidiaceihabitans, no relative Rhodobacteraceae, and Stappiaceae and by the NS4 marine group from Bacteroidota. In addition to these groups characterising only the period from April to May the gammaproteobacterial OM60 (NOR5) clade and Litoricola were more pronounced in two periods, from April to May and from June to October. Members within the Rhodobacteraceae were previously associated with phytoplankton blooms in the North Sea3,9, while the NS4 marine group was found in studies describing bacterial communities in different environments of the Adriatic Sea with no clear association with increased autotrophic biomass27,28. The observed differences between the community characteristic for the period from April to May and spring-specific communities from other areas could therefore be explained by differences in structure and supply of phototroph-derived organic matter. The community originating from the period from June to October was characterised by the AEGEAN-169 marine group, SAR11 subclade III, and HIMB11 from Alphaproteobacteria and uncultured Flavobacteriaceae, the family Balneolaceae (Balneola and uncultured Balneolaceae), and the NS11-12 marine group from Bacteroidota. As mentioned above, this period was also characterised by the the OM60 (NOR5) clade and Litoricola from Gammaproteobacteria. A higher contribution of members such as the HIMB11 and the OM60 (NOR5) clade during the warm period of the year could result from their adaptation to more oligotrophic conditions during water column stratification through the ability to use alternative pathways of energy supply, e.g. bacteriochlorophyll and proteorhodopsin32,33. The period from November to March was characterised by the alphaproteobacterial SAR11 subclade Ia, the gammaproteobacterial SUP05 cluster, and the archaeal “Candidatus Nitrosopumilus” and Marine group II. In contrast to other studies11,34,35, we observed a higher contribution of the SAR11 subclade Ia in the colder period of the year. A study describing a strong co-dominance of “Candidatus Nitrosopumilus” and Marine group II suggests that nitrification by ammonia-oxidising archaea is coupled with ammonification performed by the members of the Marine group II36. In addition, the presence of “Candidatus Nitrosopumilus” reads in our samples is not surprising as recently two new strains of ammonia-oxidising archaea within the genus Nitrosopumilus have been isolated from northern Adriatic coastal waters37.
In conclusion, prokaryotic communities inhabiting the proximity of the shore exhibit temporal variations similar to surface water assemblages in other temperate regions. A richness maximum was recorded in the colder period of the year and temporal community shifts were observed with distinct community structures characteristic for periods from April to May, June to October, and November to March. Temperature was identified as the main force driving seasonal community change. Community compositions specific for the identified time periods and taxa exhibiting temporal patterns different from other coastal areas indicate that beside global driving factors local conditions also influence the coastal prokaryotic community.
Methods
Sampling
Seawater from the northern Adriatic Sea was collected by diving (depth, 1.5 m) in the proximity of the shore (25 – 50 m distance) in two bays 7 km apart, Saline (\(45^{\circ }\)\(7^{\prime }\)\(5^{\prime \prime }\) N, \(13^{\circ }\)\(37^{\prime }\)\(20^{\prime \prime }\) E) and Funtana (\(45^{\circ }\) \(10^{\prime }\)\(39^{\prime \prime }\) N, \(13^{\circ }\)\(35^{\prime }\)\(42^{\prime \prime }\) E). The maximum depth at sampling stations was 3 – 4 m in the Bay of Saline and 2 – 2.5 m in the Bay of Funtana. Samples were collected in several 10 l containers and transported to the laboratory where 10 – 20 l were filtered through a 20 μm mesh. The filtrate was further sequentially filtered using a peristaltic pump through 3 and 0.2 μm polycarbonate membrane filters (Whatman, United Kingdom). Filters (0.2 μm) were dried briefly at room temperature and stored at −80 °C. Samples were collected monthly from July 2017 to October 2018. Concurrently of sampling for picoplankton community structure assessment, additional samples were collected to determine environmental parameters (temperature, salinity, orthophosphate, ammonium, nitrite, nitrate, silicic acid, particulate matter, chlorophyll a, and prokaryotic abundance) as reported previously17,18. Briefly, temperature and salinity were recorded on sampling dates by a pIONeer 65 probe (Radiometer Analytical, Denmark). Concentrations of orthophosphate, ammonium, nitrite, nitrate, and silicic acid were determined spectrophotometrically according to Strickland and Parsons (1972)38. Particulate matter was assessed gravimetrically by filtering up to 5 l of seawater through preweighed GF/F filters (Whatman, United Kingdom) and subsequently reweighing the filters after drying at 60 °C. Chlorophyll a was measured fluorometrically after seawater filtration through a GF/F filter (Whatman, United Kingdom) and extraction from filter in 90 % acetone39. Seawater samples for prokaryotic abundance assessment were fixed with formaldehyde (final concentration 4 %) and stained with 4,6-diamidino-2-phenylindol (DAPI, final concentration 1 μg ml−1) for 10 min40. DAPI stained samples were filtered through 0.2 μm black polycarbonate membrane filters (Whatman, United Kingdom). Abundances of prokaryotic cells were calculated after cell counting under an epifluorescence microscope (Zeiss Axio Imager Z1, Germany).
DNA extraction
Picoplankton DNA was extracted from 0.2 μm polycarbonate filters according to Massana et al. 41 with slight modifications. Following phenol-chloroform extractions, 1/10 of 3 M sodium acetate (pH 5.2) was added. DNA was precipitated by the addition of 1 volume of chilled isopropanol, by incubating the mixtures overnight at −20 °C , and by centrifuging at 20,000 \(\times\) g and 4 °C for 21 min. Pellets were washed twice with 500 μl of chilled 70 % ethanol and centrifuged after each washing step at 20,000 \(\times\) g and 4 °C for 5 min. Air-dried pellets were resuspended in 50 μl of deionized water.
Illumina 16S rRNA sequencing
The V4 region of the gene for 16S rRNA was sequenced using the Illumina MiSeq platform as described previously42. A two-step PCR procedure was applied to amplify the target region. In the first PCR, primers 515F (\(5'\)-GTGYCAGCMGCCGCGGTAA-\(3'\)) and 806R (\(5'\)-GGACTACNVGGGTWTCTAAT-\(3'\)) from the Earth Microbiome Project (https://earthmicrobiome.org/protocols-and-standards/16s) were used43,44,45. A tag sequence was added to these primers on their \(5'\) end. PCR products were purified and sent for Illumina MiSeq sequencing at IMGM Laboratories, Martinsried, Germany. Prior to sequencing at IMGM, adapter and sample-specific index sequences were incorporated during the second PCR amplification of the two-step PCR procedure using primers targeting the tagged region. Beside samples, a positive and a negative control were included in each sequencing batch. For the positive control a mock community consisting of evenly mixed DNA material originating from 20 bacterial strains (ATCC MSA-1002, ATCC, USA) was used, while the negative control comprised PCR reactions without DNA template. Reads from this study (Bay of Saline) were combined with previously reported reads obtained using the same experimental procedure (Bay of Funtana)46 and analysed together. Previously reported reads were used in a study describing the temporal dynamics of epiphytic microbial communities on marine macrophyte surfaces solely to compare the epiphytic communities with assemblages from the surrounding ambient seawater46.
Sequence and data analyses
Sequences obtained in the present study were analysed using mothur (version 1.43.0)47 according to the MiSeq Standard Operating Procedure (MiSeq SOP; https://mothur.org/wiki/MiSeq_SOP)48 and recommendations given by the Riffomonas project to enhance data reproducibility (https://riffomonas.org). Sequences were clustered into operational taxonomic units (OTUs) at a similarity level of 97 % as suggested by the MiSeq SOP. Computing was performed on the computer cluster Isabella (University Computing Centre, University of Zagreb). Alignment and classification were performed using the SILVA SSU Ref NR 99 database (release 138; https://www.arb-silva.de)49,50. The average sequencing error rate of 0.01 % was calculated based on the ATCC MSA-1002 mock community included in each sequencing batch, which is in line with previously reported values for next-generation 16S rRNA amplicon sequencing48,51. Also, negative controls processed together with the samples yielded only 2 sequences after quality curation.
Pipeline data processing and visualisation were done using R (version 4.1.1)52 in combination with packages vegan (version 2.5-7)53, stats (version 4.1.1)52, tidyverse (version 1.3.1)54, lemon (version 0.4.5)55, cowplot (version 1.1.1)56, RColorBrewer (version 1.1-2)57, kableExtra (version 1.3.4)58, rmarkdown (version 2.9)59,60,61, knitr (version 1.33)62,63,64, and tinytex (version 0.32)65,66. The observed number of OTUs, Chao1 and ACE richness estimators, and Shannon and Inverse Simpson diversity indices were calculated after normalization to the minimum number of reads per sample using vegan’s function rrarefy to account for different sequencing depths. Chao1 and ACE estimates were retrieved using vegan’s function estimateR, while Shannon and Inverse Simpson diversity indices were calculated using vegan’s function diversity53. To express both diversity indices in terms of effective number of OTUs the exponential of the Shannon diversity index was calculated67. The proportions of shared OTUs and communities between samples were expressed as the Jaccard’s (on presence/absence data) and Bray–Curtis similarity coefficient, respectively. Both coefficients were calculated on the OTU data table using vegan’s function vegdist and converted from dissimilarities to similarities53,68,69. Principal Coordinates Analysis (PCoA) was performed on Bray-Curtis dissimilarities based on OTU abundances using vegan’s function wcmdscale. The Lingoes correction method was applied to account for negative eigenvalues53,69. Difference between period-specific communities was tested by performing the Analysis of Similarities (ANOSIM) using vegan’s function anosim and 999 permutations53. Distance-based Redundancy Analysis (db-RDA) was computed by applying vegan’s function capscale to OTU abundances and explanatory environmental variables mentioned above. The analysis was performed on Bray-Curtis dissimilarities using the Lingoes correction for negative eigenvalues53,68,69. The proportion of community data variation explained by environmental variables (\(R^2_a\)) was computed using vegan’s function RsquareAdj53,68. Packages tidyverse, lemon, cowplot, and RColorBrewer were used for data analysis and visualisation, the stats package for general analysis, the help of package kableExtra to create tables, and packages rmarkdown, knitr, and tinytex to generate documents.
Data availability
The datasets generated and analysed during the current study are available in the European Nucleotide Archive (ENA) repository at EMBL-EBI under accession numbers SAMEA8117500 – SAMEA8117516, https://www.ebi.ac.uk/ena/browser/view/PRJEB43226, while the datasets analysed are available under accession numbers SAMEA6648771 – SAMEA6648788, SAMEA6648824, SAMEA6648825, https://www.ebi.ac.uk/ena/browser/view/PRJEB37267. The detailed analysis procedure including the R Markdown file are available in the GitHub repository (https://github.com/MicrobesRovinj/Korlevic_SeawaterDynamics_SciRep_2022).
References
Bunse, C. & Pinhassi, J. Marine bacterioplankton seasonal succession dynamics. Trends Microbiol. 25, 494–505. https://doi.org/10.1016/j.tim.2016.12.013 (2017).
Mestre, M., Höfer, J., Sala, M. M. & Gasol, J. M. Seasonal variation of bacterial diversity along the marine particulate matter continuum. Front. Microbiol. 11, 1590. https://doi.org/10.3389/fmicb.2020.01590 (2020).
Teeling, H. et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611. https://doi.org/10.1126/science.1218344 (2012).
Gilbert, J. A. et al. The seasonal structure of microbial communities in the Western English Channel. Environ. Microbiol. 11, 3132–3139. https://doi.org/10.1111/j.1462-2920.2009.02017.x (2009).
Sintes, E., Witte, H., Stodderegger, K., Steiner, P. & Herndl, G. J. Temporal dynamics in the free-living bacterial community composition in the coastal North Sea. FEMS Microbiol. Ecol. 83, 413–424. https://doi.org/10.1111/1574-6941.12003 (2013).
Lindh, M. V. et al. Disentangling seasonal bacterioplankton population dynamics by high-frequency sampling. Environ. Microbiol. 17, 2459–2476. https://doi.org/10.1111/1462-2920.12720 (2015).
El-Swais, H., Dunn, K. A., Bielawski, J. P., Li, W. K. W. & Walsh, D. A. Seasonal assemblages and short-lived blooms in coastal north-west Atlantic Ocean bacterioplankton. Environ. Microbiol. 17, 3642–3661. https://doi.org/10.1111/1462-2920.12629 (2015).
Ward, C. S. et al. Annual community patterns are driven by seasonal switching between closely related marine bacteria. ISME J. 11, 1412–1422. https://doi.org/10.1038/ismej.2017.4 (2017).
Teeling, H. et al. Recurring patterns in bacterioplankton dynamics during coastal spring algae blooms. Elife 5, e11888. https://doi.org/10.7554/eLife.11888 (2016).
Tinta, T. et al. Bacterial community shift is induced by dynamic environmental parameters in a changing coastal ecosystem (northern Adriatic, northeastern Mediterranean Sea) - a 2-year time-series study. Environ. Microbiol. 17, 3581–3596. https://doi.org/10.1111/1462-2920.12519 (2015).
Salter, I. et al. Seasonal dynamics of active SAR11 ecotypes in the oligotrophic Northwest Mediterranean Sea. ISME J. 9, 347–360. https://doi.org/10.1038/ismej.2014.129 (2015).
Gilbert, J. A. et al. Defining seasonal marine microbial community dynamics. ISME J. 6, 298–308. https://doi.org/10.1038/ismej.2011.107 (2012).
Alonso-Sáez, L. et al. Seasonality in bacterial diversity in north-west Mediterranean coastal waters: Assessment through clone libraries, fingerprinting and FISH. FEMS Microbiol. Ecol. 60, 98–112. https://doi.org/10.1111/j.1574-6941.2006.00276.x (2007).
Alonso-Sáez, L., Díaz-Pérez, L. & Morán, X. A. G. The hidden seasonality of the rare biosphere in coastal marine bacterioplankton. Environ. Microbiol. 17, 3766–3780. https://doi.org/10.1111/1462-2920.12801 (2015).
Needham, D. M. & Fuhrman, J. A. Pronounced daily succession of phytoplankton, archaea and bacteria following a spring bloom. Nat. Microbiol. 1, 1–7. https://doi.org/10.1038/nmicrobiol.2016.5 (2016).
Fuhrman, J. A., Cram, J. A. & Needham, D. M. Marine microbial community dynamics and their ecological interpretation. Nat. Rev. Microbiol. 13, 133–146. https://doi.org/10.1038/nrmicro3417 (2015).
Najdek, M. et al. Dynamics of environmental conditions during the decline of a Cymodocea nodosa meadow. Biogeosciences 17, 3299–3315. https://doi.org/10.5194/bg-17-3299-2020 (2020).
Najdek, M. et al. Effects of the invasion of Caulerpa cylindracea in a Cymodocea nodosa meadow in the Northern Adriatic Sea. Front. Mar. Sci. 7, 602055. https://doi.org/10.3389/fmars.2020.602055 (2020).
Ladau, J. et al. Global marine bacterial diversity peaks at high latitudes in winter. ISME J. 7, 1669–1677. https://doi.org/10.1038/ismej.2013.37 (2013).
García, F. C., Alonso-Sáez, L., Morén, X. A. G. & López-Urrutia, Á. Seasonality in molecular and cytometric diversity of marine bacterioplankton: The re-shuffling of bacterial taxa by vertical mixing. Environ. Microbiol. 17, 4133–4142. https://doi.org/10.1111/1462-2920.12984 (2015).
Reinthaler, T., Winter, C. & Herndl, G. J. Relationship between bacterioplankton richness, respiration, and production in the southern North Sea. Appl. Environ. Microbiol. 71, 2260–2266. https://doi.org/10.1128/AEM.71.5.2260-2266.2005 (2005).
Mozetič, P. et al. Recent trends towards oligotrophication of the Northern Adriatic: Evidence from chlorophyll a time series. Estuaries Coast 33, 362–375. https://doi.org/10.1007/s12237-009-9191-7 (2010).
Manna, V., De Vittor, C., Giani, M., Del Negro, P. & Celussi, M. Long-term patterns and drivers of microbial organic matter utilization in the northernmost basin of the Mediterranean Sea. Mar. Environ. Res. 164, 105245. https://doi.org/10.1016/j.marenvres.2020.105245 (2021).
Ivančić, I. et al. Long-term changes in heterotrophic prokaryotes abundance and growth characteristics in the northern Adriatic Sea. J. Mar. Syst. 82, 206–216. https://doi.org/10.1016/j.jmarsys.2010.05.008 (2010).
Bowman, J. P. The family Cryomorphaceae. In The Prokaryotes: Other Major Lineages of Bacteria and the Archaea (eds Rosenberg, E. et al.) (Springer, New York, 2014). https://doi.org/10.1007/978-3-642-38954-2_135.
Ngugi, D. K. & Stingl, U. High-quality draft single-cell genome sequence of the NS5 marine group from the coastal Red Sea. Genome Announc. 6, e00565-18. https://doi.org/10.1128/genomeA.00565-18 (2018).
Korlević, M., Pop Ristova, P., Garić, R., Amann, R. & Orlić, S. Bacterial diversity in the South Adriatic Sea during a strong, deep winter convection year. Appl. Environ. Microbiol. 81, 1715–1726; https://doi.org/10.1128/AEM.03410-14 (2015).
Korlević, M. et al. Bacterial diversity across a highly stratified ecosystem: A salt-wedge Mediterranean estuary. Syst. Appl. Microbiol. 39, 398–408. https://doi.org/10.1016/j.syapm.2016.06.006 (2016).
Hoarfrost, A. et al. Global ecotypes in the ubiquitous marine clade SAR86. ISME J. 14, 178–188. https://doi.org/10.1038/s41396-019-0516-7 (2020).
Šilović, T., Balagué, V., Orlić, S. & Pedrós-Alió, C. Picoplankton seasonal variation and community structure in the northeast Adriatic coastal zone. FEMS Microbiol. Ecol. 82, 678–691. https://doi.org/10.1111/j.1574-6941.2012.01438.x (2012).
Palenik, B. et al. The genome of a motile marine Synechococcus. Nature 424, 1037–1042. https://doi.org/10.1038/nature01943 (2003).
Spring, S. & Riedel, T. Mixotrophic growth of bacteriochlorophyll a-containing members of the OM60/NOR5 clade of marine gammaproteobacteria is carbon-starvation independent and correlates with the type of carbon source and oxygen availability. BMC Microbiol. 13, 117. https://doi.org/10.1186/1471-2180-13-117 (2013).
Durham, B. P. et al. Draft genome sequence of marine alphaproteobacterial strain HIMB11, the first cultivated representative of a unique lineage within the Roseobacter clade possessing an unusually small genome. Stand. Genomic Sci. 9, 632–645. https://doi.org/10.4056/sigs.4998989 (2014).
Carlson, C. A. et al. Seasonal dynamics of SAR11 populations in the euphotic and mesopelagic zones of the northwestern Sargasso Sea. ISME J. 3, 283–295. https://doi.org/10.1038/ismej.2008.117 (2009).
Vergin, K. L. et al. High-resolution SAR11 ecotype dynamics at the Bermuda Atlantic Time-series study site by phylogenetic placement of pyrosequences. ISME J. 7, 1322–1332. https://doi.org/10.1038/ismej.2013.32 (2013).
Kim, J.-G. et al. Distinct temporal dynamics of planktonic archaeal and bacterial assemblages in the bays of the Yellow Sea. PLoS One 14, e0221408. https://doi.org/10.1371/journal.pone.0221408 (2019).
Bayer, B. et al. Nitrosopumilus adriaticus sp. nov. and Nitrosopumilus piranensis sp. nov., two ammonia-oxidizing archaea from the Adriatic Sea and members of the class Nitrososphaeria. Int. J. Syst. Evol. Microbiol. 69, 1892–1902. https://doi.org/10.1099/ijsem.0.003360 (2019).
Strickland, J. D. H. & Parsons, T. R. A Practical Handbook of Seawater Analysis vol. 167 (Fisheries Research Board of Canada, 1972).
Holm-Hansen, O., Lorenzen, C. J., Holmes, R. W. & Strickland, J. D. H. Fluorometric determination of chlorophyll. ICES J. Mar. Sci. 30, 3–15. https://doi.org/10.1093/icesjms/30.1.3 (1965).
Porter, K. G. & Feig, Y. S. The use of DAPI for identifying and counting aquatic microflora. Limnol. Oceanogr. 25, 943–948. https://doi.org/10.4319/lo.1980.25.5.0943 (1980).
Massana, R., Murray, A. E., Preston, C. M. & DeLong, E. F. Vertical distribution and phylogenetic characterization of marine planktonic Archaea in the Santa Barbara Channel. Appl. Environ. Microbiol. 63, 50–56. https://doi.org/10.1128/aem.63.1.50-56.1997 (1997).
Korlević, M., Markovski, M., Zhao, Z., Herndl, G. J. & Najdek, M. Selective DNA and protein isolation from marine macrophyte surfaces. Front. Microbiol. 12, 665999. https://doi.org/10.3389/fmicb.2021.665999 (2021).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. https://doi.org/10.1038/ismej.2012.8 (2012).
Apprill, A., McNally, S., Parsons, R. & Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 75, 129–137. https://doi.org/10.3354/ame01753 (2015).
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414. https://doi.org/10.1111/1462-2920.13023 (2016).
Korlević, M., Markovski, M., Zhao, Z., Herndl, G. J. & Najdek, M. Seasonal dynamics of epiphytic microbial communities on marine macrophyte surfaces. Front. Microbiol. 12, 671342. https://doi.org/10.3389/fmicb.2021.671342 (2021).
Schloss, P. D. et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. https://doi.org/10.1128/AEM.01541-09 (2009).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. https://doi.org/10.1128/AEM.01043-13 (2013).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. https://doi.org/10.1093/nar/gks1219 (2013).
Yilmaz, P. et al. The SILVA and "All-species Living Tree Project (LTP)" taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. https://doi.org/10.1093/nar/gkt1209 (2014).
Schloss, P. D., Jenior, M. L., Koumpouras, C. C., Westcott, S. L. & Highlander, S. K. Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ 4, e1869. https://doi.org/10.7717/peerj.1869 (2016).
R Core Team. R: A language and environment for statistical computing (R Foundation for Statistical Computing, 2021).
Oksanen, J. et al. vegan: Community ecology package (2020).
Wickham, H. et al. Welcome to the tidyverse. J. Open Source Softw. 4, 1686; https://doi.org/10.21105/joss.01686 (2019)
McKinnon Edwards, S. lemon: Freshing up your ’ggplot2’ plots (2020).
Wilke, C. O. cowplot: Streamlined plot theme and plot annotations for ’ggplot2’ (2020).
Neuwirth, E. RColorBrewer: ColorBrewer palettes (2014).
Zhu, H. kableExtra: Construct complex table with ’kable’ and pipe syntax (2021).
Allaire, J. et al. rmarkdown: Dynamic documents for R (2021).
Xie, Y., Allaire, J. J. & Grolemund, G. R Markdown: The Definitive Guide (Chapman and Hall/CRC, New York, 2018).
Xie, Y., Dervieux, C. & Riederer, E. R Markdown Cookbook (Chapman and Hall/CRC, New York, 2020).
Xie, Y. knitr: A general-purpose package for dynamic report generation in R (2021).
Xie, Y. & knitr, A comprehensive tool for reproducible research in R. In Implementing Reproducible Computational Research (eds Stodden, V. et al.) (Chapman and Hall/CRC, New York, 2014).
Xie, Y. Dynamic Documents with R and knitr (Chapman and Hall/CRC, New York, 2015).
Xie, Y. tinytex: Helper functions to install and maintain TeX Live, and compile LaTeX documents (2021).
Xie, Y. TinyTeX: A lightweight, cross-platform, and easy-to-maintain LaTeX distribution based on TeX Live. TUGboat 40, 30–32 (2019).
Jost, L. Entropy and diversity. Oikos 113, 363–375. https://doi.org/10.1111/j.2006.0030-1299.14714.x (2006).
Borcard, D., Gillet, F. & Legendre, P. Numerical Ecology with R (Springer, New York, 2018). https://doi.org/10.1007/978-3-319-71404-2.
Legendre, P. & Legendre, L. Numerical Ecology (Elsevier, Amsterdam, 2012).
Acknowledgements
This study was funded by the Croatian Science Foundation through the MICRO-SEAGRASS project (project number IP-2016-06-7118). G.J.H. was supported by the Austrian Science Fund (FWF) through the ARTEMIS project (project number P28781-B21). We would like to thank the University Computing Centre of the University of Zagreb for access to the computer cluster Isabella, Margareta Buterer for technical support, and Paolo Paliaga for help during sampling.
Author information
Authors and Affiliations
Contributions
M.K., G.J.H., and M.N. designed the study. M.K., M.M., and M.N. performed sampling and laboratory analysis. M.K. and M.M. analysed the data. M.K. prepared the manuscript with editorial help from M.M., G.J.H., and MN. All authors contributed to the article and approved the final submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Korlević, M., Markovski, M., Herndl, G.J. et al. Temporal variation in the prokaryotic community of a nearshore marine environment. Sci Rep 12, 16859 (2022). https://doi.org/10.1038/s41598-022-20954-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-20954-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
