
Abstract
Twelve families with exuberant and early-onset calcium pyrophosphate dehydrate chondrocalcinosis (CC) and diffuse idiopathic skeletal hyperostosis (DISH), hereafter designated DISH/CC, were identified in Terceira Island, the Azores, Portugal. Ninety-two (92) individuals from these families were selected for whole-genome-wide linkage analysis. An identity-by-descent (IBD) analysis was performed in 10 individuals from 5 of the investigated pedigrees. The chromosome area with the maximal logarithm of the odds score (1.32; P=0.007) was not identified using the IBD/identity-by-state (IBS) analysis; therefore, it was not investigated further. From the IBD/IBS analysis, two candidate genes, LEMD3 and RSPO4, were identified and sequenced. Nine genetic variants were identified in the RSPO4 gene; one regulatory variant (rs146447064) was significantly more frequent in control individuals than in DISH/CC patients (P=0.03). Four variants were identified in LEMD3, and the rs201930700 variant was further investigated using segregation analysis. None of the genetic variants in RSPO4 or LEMD3 segregated within the studied families. Therefore, although a major genetic effect was shown to determine DISH/CC occurrence within these families, the specific genetic variants involved were not identified.
Similar content being viewed by others


Introduction
Diffuse idiopathic skeletal hyperostosis (DISH, MIM 106400) and chondrocalcinosis (CC, MIM 118600) are diseases characterized by ectopic calcification. DISH is characterized by the ossification of entheses in the axial and peripheral skeleton that affect the anterior spinous longitudinal ligament, in particular the right side of the spine, with preservation of the intervertebral disc space.1 CC is characterized by the deposition of calcium-containing crystals in articular cartilage, synovial membranes and, less often, in periarticular soft tissues.2,3 ANKH mutations are the only known cause of a very small number of cases of monogenic CC (MIM 118600)4–7 as well as craniometaphyseal dysplasia (MIM 123000).8–10 The ANKH gene maps to chromosome 5p15.1 and encodes the multipass transmembrane protein ANK that transports intracellular inorganic pyrophosphate to the extracellular milieu,11 where it acts as a potent inhibitor of mineralization.12 The etiology of DISH is still unknown, but several lines of evidence suggest that genetic factors might be involved in its etiology.13–15 Very few genetic studies on DISH have been published, and until now, only COL6A1 16,17 and FGF218 have been shown to have a positive association with DISH susceptibility. Moreover, all the variants in both genes that showed significant associations were located in non-coding regions and are very common variants within the general population, suggesting that these variants have a minor effect on DISH susceptibility. DISH can coexist with ossification of the posterior longitudinal ligament (MIM 602475), a disease in which the genetic background is considered relevant to its etiology. Unlike DISH, ossification of the posterior longitudinal ligament has been extensively investigated, and despite some conflicting studies, a great number of susceptibility genes have been reported. These genes include collagen 6A1 and 11A2 (COL6A1 and COL11A2, respectively),17,19 bone morphogenetic protein 2 and 4 (BMP-2 and BMP-4, respectively),20,21 ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1),22 transforming growth factor 1 and 3 receptor (TGFβ1 and TGFβ3, respectively),23,24 estrogen receptor 1 (ESR1)24 and R-spondin 2 (RSPO2)25,26 among others. However, as occurs in DISH, the genetic variants with a positive association with ossification of the posterior longitudinal ligament appear to have a minor effect on disease susceptibility, suggesting that the heritable component of its etiology is likely to be polygenic in most cases.
Genetic and clinical links exist between CC and DISH, and there is evidence that disordered pyrophosphate metabolism may have an important role; in general, conditions that favor increases in inorganic pyrophosphate promote calcium pyrophosphate dehydrate crystal formation.27 There is evidence of genetic involvement in CC and forms of spinal ossification in both human5 and animal models; the ank mouse develops severe hydroxyapatite CC and spinal ossification,11 and the tip-toe walking (ttw) mouse is the main model for human ossification of the posterior longitudinal ligament and displays cartilage calcification.28
The coexistence of DISH with CC is very common in Terceira Island—Azores, suggesting that both diseases, hereafter designated the DISH/CC phenotype, share the same pathogenic mechanism.29 A similar phenotype has also been reported in several previous studies.30,31 Our main objective was to elucidate the genetic factors underlying this condition and to establish whether this disease is a novel form of pyrophosphate arthropathy or a simple association between two different disorders with extremely high prevalence on this island in the Azores. A formal segregation study in these families suggested that the condition was caused by a Mendelian inherited disorder with an autosomal-dominant model of transmission.32 Therefore, we performed a whole-genome linkage analysis followed by ‘identity-by-state (IBS)/identity-by-descent (IBD)’ mapping to determine the genetic cause of this phenotype. IBD mapping is a statistical method for the detection of genetic loci that share an ancestral segment among ‘unrelated’ affected pairs of individuals. IBD mapping is a more robust approach than allelic heterogeneity and can be used as a complementary method to genome-wide linkage studies to identify rare inherited variants when combined with sequencing data.33
Materials and methods
Collections used
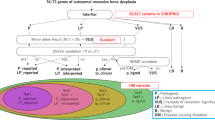
Twelve pedigrees with 92 individuals were used for the linkage analysis.
Ten affected individuals from five pedigrees previously used for linkage analysis were selected for IBD/IBS analysis. The radiological characterization of these individuals is presented in Table 1.
This study was approved by the HSEIT Ethics Committee and all participants provided informed consent. Peripheral blood was collected for biochemical analysis and DNA isolation.
Microsatellite genotyping
Highly informative microsatellite markers from the Linkage Mapping Set V2 (Applied Biosystems, Foster City, CA, USA) were used for the linkage analysis. Microsatellites were amplified in 20 μl PCR reactions using optimized reaction conditions. To simplify the genotyping process, PCR products were pooled based on the fluorochrome used, the allele-size range and avoiding fluorochromes with overlapping spectra or overlapping alleles. The 96-well plates were prepared by adding a mixture of 10 μl of Hi-Di formamide (Applied Biosystems) and 0.3 μl of the internal line standard GeneScan 400HD-ROX (Applied Biosystems) to each well. Samples were denatured and immediately run on an ABI automated DNA genetic analyzer 3700 and 3130XL. Microsatellite genotype calling was performed semiautomatically using the GENOTYPER software (Applied Biosystems) and the GeneMapper Software Version 4.0 (Applied Biosystems).
Quality control and linkage analysis
To minimize data errors, extensive checking procedures were undertaken. Genetic Analysis System (GAS version 2.0; Alan Young, Oxford University, Oxford, UK) was used to assign discrete allele numbers to the alleles. Mendelian inheritance was checked with GENOTYPER and GeneMapper (Applied Biosystems). PEDCHECK34 was used to screen all the data for previously undetected Mendelian inconsistencies. Statistical analysis and parametric and non-parametric analyses were performed using VITESSSE,35 GENEHUNTER and MERLIN, respectively.36 Marker positions were obtained from the National Center for Biotechnology Information database (NCBI). Parametric linkage analysis was performed assuming autosomal-dominant transmission, a penetrance of 90–95%, phenocopy rates of 0.05–0.1% and minor allele frequencies of 0.1–1%.
IBD/IBS analysis
Genotyping was performed with Illumina (San Diego, CA, USA) 370CNV BeadChips. To perform the analysis, 50,000 markers with the following characteristics were chosen: (a) not in a linkage disequilibrium with each other (r2<0.1) and (b) had a minor allele frequency >0.3. These were chosen using the TASCOG control set (approximately 400 individuals with predominantly UK ancestries from Tasmania, Australia).37 IBD segments were then inferred for all pairs of cases across the genome using PLINK38 with the ‘Spairs’ statistic, which represents the number of pairs of cases that appear to have IBD inherited chromosomal segments at each point on the genome.
Using the fastIBD option in the software package BEAGLE (2),39 it was possible to identify IBD regions across families while also taking into account the linkage disequilibrium. The analysis was set for a minimum IBD segment length of 1 cM and an IBD detection threshold of 10−10. The genome-wide P-value was estimated knowing that there were 366 segments with an average of 3.1 case pairs shared across the genome. The number of case pairs shared across the genome approximately followed a Poisson distribution. As the 366 segments were not independent across the genome, principal component analysis was used to show that the 366 multiple tests performed were approximately equivalent to 202 independent tests.40 The genome wide P-value was obtained by estimating the probability of obtaining a maximum number of case pairs shared out of 202 draws from the Poisson distribution (lambda=3.1) by chance. As a maximum of 10 cases were observed to share IBD, this probability was estimated to be 0.077 by assuming that the number of case pairs shared had a Poisson distribution with a mean of 3.1.
Sanger sequencing
RSPO4 gene primer pairs were designed using the Primer3 software (Open source developed and maintained by Whitehead Institute for Biomedical Research, Cambridge, MA, USA). Regulatory coding regions, including intron–exon boundaries, were amplified and sequenced in 55 patients with DISH/CC disease and 36 unaffected control subjects. The LEMD3 gene was sequenced using the primers previously described by Hellemans et al.41 The amplification conditions for all the primers are available upon request. PCR fragments were purified with ExoSAP-IT (Exonuclease I and Shrimp Alkaline Phosphatase) and sequenced using ABI Big Dye chemistry (unidirectional or, when necessary, bidirectional) followed by purification with EDTA/sodium acetate and ethanol precipitation. Sequencing products were run on an automated DNA sequencer (ABI 3130XL, Applied Biosystems), and genetic variants were screened by sequencing analysis with SeqScape (Applied Biosystems). Base calling for heterozygous positions occurred when the smaller peak of two co-incident peaks was >25% of the maximum peak.
Whenever possible, the genetic variants found in the RSPO4 gene were screened across all family members when an affected individual was identified. The LEMD3 rs201930700 variant was typed in a representative randomized group of 124 individuals from Terceira Island.
Statistical analysis
The Hardy–Weinberg equilibrium and all other statistical analyses were performed using the PLINK software.38 Association was tested between the DISH/CC disease and allelic variants with the Cochran–Armitage trending test, dominant and recessive gene action tests with 1 degree of freedom and a genotypic test with two degrees of freedom. The Fisher’s exact test was used to assess the differences in the allele frequencies between the 55 patients with DISH/CC and the 36 control individuals. For all statistical tests, a P-value⩽0.05 was considered statistically significant.
Results
Linkage analysis
A whole-genome linkage scan, including 92 individuals from 12 pedigrees, was performed on all autosomes, with 189 microsatellite markers and an average distance between markers of 17 cM. Parametric and non-parametric analyses were also performed. The best results were obtained with a non-parametric linkage analysis for chromosome 16 (single point 2.38 and multipoint 1.16). Because the average distance between markers was >10 cM, one extra set of 137 markers, which was distributed on all autosomes, was further amplified and reanalyzed to enlarge the power of the study. A total of 327 microsatellites were included in the analysis. Owing to its complexity, pedigree 2 was excluded from the analysis because of computation time and memory constraints. When assuming an autosomal-dominant mode of inheritance with a minor allele frequency of 0.1%, phenocopy rate of 0.1% and penetrance of 90%, the parametric analysis did not give significant results.
Because there was only a nominally suggestive linkage to an area on chromosome 16, a new set of 13 microsatellite markers that covered this entire chromosome with an average spacing of 8.99 cM was further amplified and gave more significant results (Table 2).
The most significant result on chromosome 16 was obtained for the D16S3100 marker; it had a logarithm of the odds score of 1.32 and a P-value of 0.007. The chromosomal area (16p12.1) directly surrounding marker D16S3100 in the 26.020–27.120 K region had only 12 annotated genes, among which 6 were uncharacterized, 3 were pseudogenes and 1 microRNA and 1 open reading frame were also identified. According to the present knowledge, the only gene of putative interest in this region was HS3ST4, which encodes the enzyme heparin sulfate (glucosamine) 3-O-sulfotransferase. This region was not further investigated owing to a lack of replication using IBD analysis.
IBD sharing
There were a total of 10 cases corresponding to 45 pairs of cases in the IBD analysis. The greatest number of case pairs shared was 10, which was on chromosome 12. The 10 pairs were shared by 3 families. The significance of the association with chromosome 12 included a single-point P-value=0.0014 and a genome-wide P-value=0.077.
When using BEAGLE on the CC data set with a minimum IBD segment length of 1 cM and an IBD detection threshold of 10−10, regions on chromosomes 12 and 20 revealed the sharing of 10 pairs (Table 3). The IBD analysis did not replicate the weak suggestive linkage to an area surrounding the D16S3100 marker on chromosome 16.
The chromosome regions considered to be of interest were selected considering the maximum number of pairs shared. Two zones, one in chromosome 12 and one in chromosome 20, had a maximum of 10 pairs shared and were therefore further investigated. In contrast with previous reports that identified ANKH as a CC-associated gene, it was not identified on chromosome 5 in our study.
A total of 167 previously characterized genes were identified in the regions of interest on chromosomes 12 and 20. There were 52 genes in the region on chromosome 12 (65667554–68670915), of which only 35 were annotated. Just next to this region in chromosome 12 (65169571–652483279), one gene that had already been associated with disordered calcification, LEMD3, was identified. This gene encodes a LEM domain-containing protein. The encoded protein functions to antagonize TGF-β and BMP signaling at the inner nuclear membrane. Mutations in this gene have been associated with osteopoikilosis (MIM 166 700), Buschke–Ollendorff syndrome (MIM 166700) and melorheostosis (MIM 155950).41
There were 148 genes in chromosome 20 (chromosome region from 821749 to 6074302), of which 115 were annotated. The best candidate gene in chromosome 20 was RSPO4 (chromosome 20:958452–1002284). The RSPO4 gene encodes a member of the R-spondin family of proteins that share a common domain organization consisting of a signal peptide, cysteine-rich/furin-like domain, thrombospondin domain and a C-terminal basic region. The encoded protein may be involved in the activation of the Wnt/β-catenin signaling pathways.42 WNT signaling is critical for nail development and mutations in different WNT-associated genes have been identified in disorders involving the nails. RSPO4 mutations have been particularly associated with autosomal-recessive congenital anonychia (NDNC4; MIM 206800).43,44
Sequencing results
RSPO4 sequencing
Nine genetic variants were identified in the RSPO4 gene, including three missense variants (rs6140807 and rs61740632, rs201485021), one splice site variant (rs775644973), two synonymous (rs150446609 and rs41275604) and three regulatory region variants (rs146447064, rs149154047 and rs6056520) (Table 4). All of them were in Hardy–Weinberg equilibrium.
Two variants in the regulatory region of the RSPO4 gene (rs146447064 and rs149154047) were located in a fully conserved region and are rare (Table 4). The same was observed for the synonymous variant rs150446609, which was very rare (<0.01) and was located in a fully conserved chromosome region. The missense variant rs201485021 located in exon 3 has a SIFT score of 0 and a PolyPhen value of 1, indicating that it has deleterious and damaging effects on the protein, respectively. Additionally, this variant was located in a fully conserved region and was extremely rare (minor allele frequency <0.01); in the ‘1000 genomes’ (genomes from 26 different populations), the variant was identified in only 2 males from an Iberian population in Spain. In our study, this variant was found in one female in our control group (n=36). The other two missense variants (rs6140807 and rs61740632) had SIFT and PolyPhen values indicative of minor effects on the protein (tolerated and benign, respectively) and were both relatively rare. We found an extremely rare HGMD mutation (rs775644973 or CS065613) that has previously been associated with congenital anonychia.44 The variant in heterozygosity was found in 1 asymptomatic female in our group of 55 DISH/CC patients.
LEMD3 sequencing
LEMD3 was sequenced in the four probands from the families who shared the maximum number of pairs in the IBD/IBS analysis. The identified genetic variants can be observed in Table 5.
Variant rs201930700 was located in exon 13 of the LEMD3 gene and had a SIFT score of 0 and a PolyPhen value of 0.995, which indicated that this variant has deleterious and damaging effects on the protein, respectively. The variant was extremely rare. The minor allele frequency was unknown because there was insufficient data to establish population frequency; it was identified in only 5 of the 121,412 alleles (ExAc_Aggregated_Populations), indicating that it was unquestionably rare (minor allele frequency 0.00004). A cohort of 124 individuals, which is representative of the Terceira Island population, was typed for this mutation, and 1 other individual carrying the variant was identified. This individual and their relatives were examined (interviewed for clinical purposes, X-rayed and typed).
The rs201930700 variant was in Hardy–Weinberg equilibrium. Of the five individuals studied in the AZ3 family, four individuals (two males and two females) were affected by DISH/CC and were carriers of the rs201930700 variant and one individual (male) was affected by DISH/CC but was a non-carrier of the variant. In the AZ3 family, it was impossible to verify segregation as all the individuals studied were affected by DISH/CC affected; thus an association test was not performed.
Segregation analysis
Segregation analysis involved typing eight genetic variants in the seven DISH/CC pedigrees where the variants were found, including the seven variants in RSPO4 (rs146447064, rs149154047, rs6056520, rs150446609, rs6140807, rs41275604 and rs61740632) and variant rs201930700 in LEMD3.
Most of the analyzed families were uninformative because of the large number of affected versus unaffected individuals. Variants did not segregate within the investigated informative families (see Supplementary Figure 1 and Supplementary Table 1 for more information).
Association study
The RSPO4 regulatory region variant rs146447064 was significantly (P=0.03) more frequent in the controls (14%) than in the DISH/CC patients (5%), and when adjusted for gender, it was significant in females (P=0.0333) but not in males (Table 6).
Similar results were obtained using the Cochran–Armitage trend and allelic tests (see Supplementary Table 2). The other RSPO4 regulatory region variant, rs149154047, was also significantly different in frequency between the DISH/CC males (3%) and control males (14%) (P=0.0502) but not in females. Similar results were obtained using the Cochran–Armitage trend test (P=0.02453, CHISQ=5.057, Df=1) and the allelic test (P=0.02958, CHISQ=4.734, Df=1) (see Supplementary Table 2).
Discussion
DISH/CC is a poorly understood phenotype characterized by peripheral and axial enthesopathic calcifications, which fulfill the radiological criteria for DISH and are associated with calcium pyrophosphate dehydrate CC in some cases.31,45,46 The variable clinical and radiological presentation of patients with DISH/CC in the families investigated raises the possibility of genetic heterogeneity. The whole-genome linkage analysis found no chromosomal region with clear linkage in the affected families. The following reasons can be postulated to explain these results: (1) the phenotypic heterogeneity observed within and among pedigrees may result from genetic heterogeneity; (2) unknown environmental factors may be affecting the phenotypes and causing difficulties in the analysis; and (3) the condition may not be a simple Mendelian disorder caused by a large effect rare variant but instead may have a more complex and polygenic genetic architecture. The region on chromosome 16 that showed weak suggestive linkage was not replicated by the IBD/IBS study and was not further investigated.
The concurrence of DISH and CC suggests a shared pathogenic mechanism.32 The etiology of DISH/CC is unknown. However, because it is a bone-forming disease, it is expected that genes related to the calcification and ossification process are implicated in its etiology. In the second strategy utilized, the IBS/IBD analysis, two chromosomal areas were identified and further investigations performed. Two genes, RSPO4 and LEMD3, were selected as good candidates based on their functions. Two variants in the RSPO4 gene (rs146447064 and rs14915407), which were significantly more frequent in the controls than in DISH/CC patients, may have a possible protective role in the DISH/CC phenotype, though the level of significance observed was not definitive.
As already mentioned, the RSPO4 protein is known to have a major role in activating the Wnt/β-catenin signaling pathways.42 It is known that induction of the Wnt signaling pathway promotes bone formation, while inactivation of this pathway leads to an osteopenia state.47 Based on this knowledge, we hypothesized that rs146447064 and rs14915407 were variants associated with a reduction in RSPO4 gene expression, thus reducing Wnt activation and consequently protecting against new bone formation. Obviously, further studies are needed to test this theory.
The LEMD3 gene missense variant (rs201930700) was found in two apparently unrelated families. This variant has been identified very few times previously and both families in which it was typed presented the DISH/CC phenotype. The variant identified caused the substitution of amino acid 901 from a large and basic arginine to a large and aromatic tryptophan. Phylogenetically, according to the Ensembl database, the modified nucleotide was highly conserved in all vertebrates and was located in the carboxy-terminal nucleoplasmic region of the MAN1 protein. This region (amino acids 782–911) is predicted to be an RNA recognition motif-like (RRM-like) protein interaction domain called the U2AF homology motif.48,49 The conserved region that contains the UHM domain (U2AF homology motif kinase 1) is exclusive to MAN1 proteins and is essential for smad2 and smad3 binding.50 It is unknown whether interactions between MAN1 and Smad1 or Smad2 and Smad3 inhibit BMP and TGF-β signaling, respectively.51,52 It has been reported that heterozygous loss-of-function mutations in LEMD3 enhance TGF-β signaling and lead to sclerosing bone dysplasia, osteopoikilosis and Buschke–Ollendorff syndrome.41 As the identified variant (rs201930700) was deleterious but was not a loss-of-function mutation, the carriers of the variant did not present any signs of osteopoikilosis or Buschke–Ollendorff Syndrome. At this point, it was difficult to ascertain the effect produced on the DISH/CC phenotype by the rs201930700 variant. We postulate that the rs201930700 variant may promote enhanced TGF-β signaling, leading to increased bone formation. Our hypothesis was not confirmed by the segregation analysis, which might be due to the characteristics of the pedigrees used, as almost all the individuals were DISH/CC affected, making it very difficult to verify segregation. Other studies will be necessary to verify the impact of this rare variant on the phenotype to establish a possible association.
In conclusion, a combined strategy led us to investigate two candidate genes, RSPO4 and LEMD3, and the results obtained suggested a small and protective role for the two RSPO4 gene regulatory variants, probably by altering RSPO4 gene expression. At this point, it is impossible to ascertain the relevance of the extremely rare variant in LEMD3 (rs201930700) to the DISH/CC phenotype. Although the results are interesting, future studies are required to confirm and assess their relevance for DISH/CC pathologies.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Mader R . Clinical manifestations of diffuse idiopathic skeletal hyperostosis of the cervical spine. Semin Arthritis Rheum 2002; 32: 130–135.
Altman RD, Muniz OE, Pita JC, Howell DS . Articular chondrocalcinosis. Microanalysis of pyrophosphate (PPi) in synovial fluid and plasma. Arthritis Rheum 1973; 16: 171–178.
Silcox DC, McCartyJr DJ . Elevated inorganic concentrations in synovial fluids in osteoarthritis and pseudogout. J Lab Clin Med 1974; 83: 518–531.
Pendleton A, Johnson MD, Hughes A, Gurley KA, Ho AM, Doherty M et al. Mutations in ANKH cause chondrocalcinosis. Am J Hum Genet 2002; 71: 933–940.
Williams CJ, Zhang Y, Timms A, Bonavita G, Caeiro F, Broxholme J et al. Autosomal dominant familial calcium pyrophosphate dihydrate deposition disease is caused by mutation in the transmembrane protein ANKH. Am J Hum Genet 2002; 71: 985–991.
Williams CJ, Pendleton A, Bonavita G, Reginato AJ, Hughes AE, Peariso S et al. Mutations in the amino terminus of ANKH in two US families with calcium pyrophosphate dihydrate crystal deposition disease. Arthritis Rheum 2003; 48: 2627–2631.
Gruber BL, Couto AR, Armas JB, Brown MA, Finzel K, Terkeltaub RA . Novel ANKH amino terminus mutation (Pro5Ser) associated with early-onset calcium pyrophosphate disease with associated phosphaturia. J Clin Rheumatol 2012; 18: 192–195.
Nurnberg P, Thiele H, Chandler D, Hohne W, Cunningham ML, Ritter H et al. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet 2001; 28: 37–41.
Kornak U, Brancati F, Le Merrer M, Lichtenbelt K, Hohne W, Tinschert S et al. Three novel mutations in the ANK membrane protein cause craniometaphyseal dysplasia with variable conductive hearing loss. Am J Med Genet A 2010; 152A: 870–874.
Dutra EH, Chen IP, McGregor TL, Ranells JD, Reichenberger EJ . Two novel large ANKH deletion mutations in sporadic cases with craniometaphyseal dysplasia. Clin Genet 2012; 81: 93–95.
Ho AM, Johnson MD, Kingsley DM . Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000; 289: 265–270.
Chen IP, Wang L, Jiang X, Aguila HL, Reichenberger EJ . A Phe377del mutation in ANK leads to impaired osteoblastogenesis and osteoclastogenesis in a mouse model for craniometaphyseal dysplasia (CMD). Hum Mol Genet 2011; 20: 948–961.
Beardwell A . Familial ankylosing vertebral hyperostosis with tylosis. Ann Rheum Dis 1969; 28: 518–523.
Woodard JC, Poulos PW Jr., Parker RB, Jackson RI Jr., Eurell JC . Canine diffuse idiopathic skeletal hyperostosis. Vet Pathol 1985; 22: 317–326.
Morgan J, Stavenborn M . Disseminated idiopathic skeletal hyperostosis (DISH) in a dog. Vet Radiol Ultrasound 1991; 32: 65–70.
Tanaka T, Ikari K, Furushima K, Okada A, Tanaka H, Furukawa K et al. Genomewide linkage and linkage disequilibrium analyses identify COL6A1, on chromosome 21, as the locus for ossification of the posterior longitudinal ligament of the spine. Am J Hum Genet 2003; 73: 812–822.
Kong Q, Ma X, Li F, Guo Z, Qi Q, Li W et al. COL6A1 polymorphisms associated with ossification of the ligamentum flavum and ossification of the posterior longitudinal ligament. Spine (Phila Pa 1976) 2007; 32: 2834–2838.
Jun JK, Kim SM . Association study of fibroblast growth factor 2 and fibroblast growth factor receptors gene polymorphism in Korean ossification of the posterior longitudinal ligament patients. J Korean Neurosurg Soc 2012; 52: 7–13.
Maeda S, Ishidou Y, Koga H, Taketomi E, Ikari K, Komiya S et al. Functional impact of human collagen alpha2(XI) gene polymorphism in pathogenesis of ossification of the posterior longitudinal ligament of the spine. J Bone Miner Res 2001; 16: 948–957.
Yan L, Chang Z, Liu Y, Li YB, He BR, Hao DJ . A single nucleotide polymorphism in the human bone morphogenetic protein-2 gene (109T>G) affects the Smad signaling pathway and the predisposition to ossification of the posterior longitudinal ligament of the spine. Chin Med J (Engl) 2013; 126: 1112–1118.
Ren Y, Feng J, Liu ZZ, Wan H, Li JH, Lin X . A new haplotype in BMP4 implicated in ossification of the posterior longitudinal ligament (OPLL) in a Chinese population. J Orthop Res 2012; 30: 748–756.
He Z, Zhu H, Ding L, Xiao H, Chen D, Xue F . Association of NPP1 polymorphism with postoperative progression of ossification of the posterior longitudinal ligament in Chinese patients. Genet Mol Res 2013; 12: 4648–4655.
Kamiya M, Harada A, Mizuno M, Iwata H, Yamada Y . Association between a polymorphism of the transforming growth factor-beta1 gene and genetic susceptibility to ossification of the posterior longitudinal ligament in Japanese patients. Spine (Phila Pa 1976) 2001; 26: 1264–1266, discussion 1266–1267.
Horikoshi T, Maeda K, Kawaguchi Y, Chiba K, Mori K, Koshizuka Y et al. A large-scale genetic association study of ossification of the posterior longitudinal ligament of the spine. Hum Genet 2006; 119: 611–616.
Nakajima M, Takahashi A, Tsuji T, Karasugi T, Baba H, Uchida K et al. A genome-wide association study identifies susceptibility loci for ossification of the posterior longitudinal ligament of the spine. Nat Genet 2014; 46: 1012–1016.
Nakajima M, Kou I, Ohashi H, Ikegawa S . Identification and functional characterization of RSPO2 as a susceptibility gene for ossification of the posterior longitudinal ligament of the spine. Am J Hum Genet 2016; 99: 202–207.
Timms AE, Zhang Y, Russel RG, Brown MA . Genetic studies of disorders of calcium crystal deposition. Rheumatol 2002; 41: 725–729.
Okawa A, Ikegawa S, Nakamura I, Goto S, Moriya H, Nakamura Y . Mapping of a gene responsible for twy (tip-toe walking Yoshimura), a mouse model of ossification of the poserior longitudinal ligament of the spine (OPLL). Mamm Genome 1998; 2: 155–156.
Bruges-Armas JB, Couto AR, Timms A, Santos MR, B.F. B, Peixoto MJ et al. Ectopic calcification in the Azores—clinical and radiological manifestations of DISH and chondrocalcinosis families. Arthritis Rheum 2006; 54: 1340–1349.
van der Korst JK, Geerards J, Driessens FC . A hereditary type of idiopathic articular chondrocalcinosis. Survey of a pedigree. Am J Med 1974; 56: 307–314.
Okazaki T, Saito T, Mitomo T, Siota Y . Pseudogout: clinical observations and chemical analyses of deposits. Arthritis Rheum 1976; 19 (Suppl 3): 293–305.
Bruges-Armas J, Couto AR, Timms A, Santos MR, Bettencourt BF, Peixoto MJ et al. Ectopic calcification among families in the Azores: clinical and radiologic manifestations in families with diffuse idiopathic skeletal hyperostosis and chondrocalcinosis. Arthritis Rheum 2006; 54: 1340–1349.
Browning SR, Thompson EA . Detecting rare variant associations by identity-by-descent mapping in case-control studies. Genetics 2012; 190: 1521–1531.
O'Connell JR, Weeks DE . PedCheck: a program for identifying genotype incompatibilities in linkage analysis. Am J Hum Genet 1997; 63: 259–266.
O'Connell JR, Weeks DE . The VITESSE algorithm for rapid exact multilocus linkage analysis via genotype set-recoding and fuzzy inheritance. Nat Genet 1995; 11: 402–408.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR . Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Srikanth V, Beare R, Blizzard L, Phan T, Stapleton J, Chen J et al. Cerebral white matter lesions, gait, and the risk of incident falls: a prospective population-based study. Stroke 2009; 40: 175–180.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Browning SR, Browning BL . Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 2007; 81: 1084–1097.
Gao X, Starmer J, Martin ER . A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet Epidemiol 2008; 32: 361–369.
Hellemans J, Preobrazhenska O, Willaert A, Debeer P, Verdonk PC, Costa T et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat Genet 2004; 36: 1213–1218.
Kim KA, Zhao J, Andarmani S, Kakitani M, Oshima T, Binnerts ME et al. R-Spondin proteins: a novel link to beta-catenin activation. Cell Cycle 2006; 5: 23–26.
Bergmann C, Senderek J, Anhuf D, Thiel CT, Ekici AB, Poblete-Gutierrez P et al. Mutations in the gene encoding the Wnt-signaling component R-spondin 4 (RSPO4) cause autosomal recessive anonychia. Am J Hum Genet 2006; 79: 1105–1109.
Blaydon DC, Ishii Y, O'Toole EA, Unsworth HC, Teh MT, Ruschendorf F et al. The gene encoding R-spondin 4 (RSPO4), a secreted protein implicated in Wnt signaling, is mutated in inherited anonychia. Nat Genet 2006; 38: 1245–1247.
van der Korst JK, Geerards J . Articular chondrocalcinosis in a Dutch pedigree. Arthritis Rheum 1976; 19 (Suppl 3): 405–409.
Harris J, Carter AR, Glick EN, Storey GO . Ankylosing hyperostosis. I. Clinical and radiological features. Ann Rheum Dis 1974; 33: 210–215.
Kim JH, Liu X, Wang J, Chen X, Zhang H, Kim SH et al. Wnt signaling in bone formation and its therapeutic potential for bone diseases. Ther Adv Musculoskelet Dis 2013; 5: 13–31.
Kielkopf CL, Lucke S, Green MR . U2AF homology motifs: protein recognition in the RRM world. Genes Dev 2004; 18: 1513–1526.
Caputo S, Couprie J, Duband-Goulet I, Konde E, Lin F, Braud S et al. The carboxyl-terminal nucleoplasmic region of MAN1 exhibits a DNA binding winged helix domain. J Biol Chem 2006; 281: 18208–18215.
Konde E, Bourgeois B, Tellier-Lebegue C, Wu W, Perez J, Caputo S et al. Structural analysis of the Smad2-MAN1 interaction that regulates transforming growth factor-beta signaling at the inner nuclear membrane. Biochemistry 2010; 49: 8020–8032.
Raju GP, Dimova N, Klein PS, Huang HC . SANE, a novel LEM domain protein, regulates bone morphogenetic protein signaling through interaction with Smad1. J Biol Chem 2003; 278: 428–437.
Lin F, Morrison JM, Wu W, Worman HJ . MAN1, an integral protein of the inner nuclear membrane, binds Smad2 and Smad3 and antagonizes transforming growth factor-beta signaling. Hum Mol Genet 2005; 14: 437–445.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Couto, A., Parreira, B., Thomson, R. et al. Combined approach for finding susceptibility genes in DISH/chondrocalcinosis families: whole-genome-wide linkage and IBS/IBD studies. Hum Genome Var 4, 17041 (2017). https://doi.org/10.1038/hgv.2017.41
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.41
