
Abstract
In an effort to identify rare alleles associated with adolescent idiopathic scoliosis (AIS) whole-exome sequencing was performed on a discovery cohort of 73 unrelated patients and 70 age-and sex matched controls, all of French-Canadian ancestry. A collapsing gene burden test was performed to analyze rare protein-altering variants using case–control statistics. Since no single gene achieved statistical significance, targeted exon sequencing was performed for 24 genes with the smallest p values, in an independent replication cohort of unrelated severely affected females with AIS and sex-matched controls (N = 96 each). An excess of rare, potentially protein-altering variants was noted in one particular gene, FAT3, although it did not achieve statistical significance. Independently, we sequenced the exomes of all members of a rare multiplex family of three affected sisters and unaffected parents. All three sisters were compound heterozygous for two rare protein-altering variants in FAT3. The parents were single heterozygotes for each variant. The two variants in the family were also present in our discovery cohort. A second validation step was done, using another independent replication cohort of 258 unrelated AIS patients having reach their skeletal maturity and 143 healthy controls to genotype nine FAT3 gene variants, including the two variants previously identified in the multiplex family: p.L517S (rs139595720) and p.L4544F (rs187159256). Interestingly, two FAT3 variants, rs139595720 (genotype A/G) and rs80293525 (genotype C/T), were enriched in severe scoliosis cases (4.5% and 2.7% respectively) compared to milder cases (1.4% and 0.7%) and healthy controls (1.6% and 0.8%). Our results implicate FAT3 as a new candidate gene in the etiology of AIS.
Similar content being viewed by others

Introduction
Adolescent Idiopathic Scoliosis (AIS) is a complex disorder of the spine, and the most common form of such disorders. It is a three-dimensional deformity of the spine characterized by a lateral curvature of ≥ 10° on a standing radiograph (Cobb method), combined with vertebral rotation. It mostly occurs at the age of adolescence and affects 1–4%1 of the global pediatric population with higher prevalence in females who are generally more severely affected than males2. In most cases the underlying cause of idiopathic scoliosis is unknown, although a genetic component is well recognized3,4. Twin and family studies have documented high rates of concordance among twins and increased risk to relatives of patients with AIS5,6. The mode of inheritance is still unclear7. The genetic nature of the disease is complex, with an apparent high level of heterogeneity between different families8,9,10. A number of candidate genes and loci have been suggested by different studies, but few have been successfully replicated11. Human genetic studies have used both linkage and association methods. The results of linkage studies have been poorly reproducible11. Genome wide association studies (GWAS) have identified several candidate genes for AIS including CHL1, LBX1, GPR126, BNC2, and PAX112,13,14,15,16. The associated common single nucleotide polymorphisms (SNPs) identified to date only explain a small portion of the genetic component of the disease. Genetic interactions17 and rare variants18 might explain part of this “missing heritability” in AIS19.
Few studies have attempted to detect rare causal variants in AIS and this field of research is still in its infancy. Sequencing using either whole exome or targeted gene panels, has identified several genes that might contribute to the occurrence and or severity of scoliosis; such as FBN1, FBN220, HSPG221, POC522, and AKAP223. Another study suggested that accumulation of rare variants in a group of genes of the extracellular matrix might contribute to disease risk24. In summary, genome-wide association studies (GWAS) cannot reveal all genetic determinants associated with AIS, which is true with other complex traits. Such limitation is not exclusive to GWAS, as no method or technology to date can identify all the genetic components of complex traits despite the fact that candidate gene approach tends to have greater statistical power than studies that use large numbers of single nucleotide. Overall, this explains why the genetic component of AIS is not yet fully understood, leaving significant room for further research.
In this study, we performed whole-exome sequencing (WES) in a French-Canadian AIS cohort, followed by a targeted sequencing of the 24 statistically-strongest candidate genes from WES, in an independent replication cohort. In parallel, we performed WES in a unique multiplex family of three affected sisters with healthy parents. Our goal was to identify new genes enriched with rare variants, which might contribute to the disease. Our results implicate a novel gene, FAT3, not previously associated with AIS, as a strong candidate for this condition.
Results
Study populations
Our discovery cohort includes 73 unrelated AIS patients (68 females and 5 males), and 70 sex- and age-matched controls, all of French-Canadian ancestry (Table 1). Fifty of the patients were considered severely affected as their Cobb angles were at least 40°, and the remaining patients were considered as moderate cases (10°–39°). Our first independent replication cohort includes 96 unrelated AIS patients (only females) and 96 healthy controls (only females), which we used for the replication of the top 24 genes from the discovery cohort (Table 2). Our second replication cohort includes 258 unrelated AIS patients (82.9% females), who have reached their skeletal maturity and stratified by spinal deformity severity (Cobb angle ≥ 40° versus Cobb angle < 40°), and 143 healthy controls (Table 3).
Whole-exome sequencing (WES)
We performed WES using our discovery cohort, followed by variant annotation and filtering to identify rare variants contributing to AIS. To enhance statistical power, we examined genes harboring an overall excess of rare variants in the discovery patient cohort. We performed a collapsing gene burden test, in which we compared the enrichment of rare variants per gene in patients versus controls. To define rare variants, we applied a minor allele frequency (MAF) < 1% as an initial cutoff, and MAF < 0.5% as a more stringent cutoff according to the 1000 Genomes Project European ancestry (EUR) and the Exome Sequencing Project European ancestry (ESP-EA). Only 8150 genes harbored at least one such rare variant among all case and control samples. Therefore, we set a statistical significance threshold at 0.05/8150 = 6 × 10−6. Based on our results, none of the 8150 genes met the p-value threshold. We therefore selected the top 24 genes with the strongest statistical scores, for follow-up validation in an independent replication cohort (Table 4). To selection the 24 candidate genes, we took into consideration both the p-values and the absolutes numbers of patients and controls who carried the rare variants.
Targeted sequencing of the selected 24 genes in a first replication cohort
The first replication cohort was chosen to be more homogeneous; all cases were severely affected females. By comparison, in our initial discovery cohort, 93% of AIS patients were females and only 68% were severe cases. This replication cohort includes 96 female patients and 96 female controls. The exons of the 24 genes were sequenced in the 192-replication samples using a custom capture library. After calling and annotating variants, we first removed poor quality calls, variants with a MAF ≥ 1% (according to the 1000 Genomes Project EUR and the gnomAD whole-genome and whole-exome databases), and synonymous variants. We included near-intronic variants that might affect efficiency of RNA splicing. Similarly, we employed the collapsing gene burden test. Of the 24 genes, only one gene, FAT3, continued to show an enrichment of rare, potentially protein-altering variants in patients versus controls. Specifically, there were 21 rare variants in FAT3, compared to 11 in controls (uncorrected p value = 0.04, Fisher-exact one-tailed test) (Table 5). We suggest that a one-tailed-test is appropriate since the primary ascertainment was for AIS cases, and rare variants in our cohorts would not realistically be expected to be protective. The p value of 0.04 is before correcting for multiple genes in the replication thus is not formally statistically significant although it is highly suggestive. Importantly, we explored other models, such as using a 2% MAF or even 5% MAF threshold instead of 1%, or filtering to retain variants with a REVEL pathogenicity score above 0.3 (the value for which REVEL specificity and sensitivity are approximately equal). The total number of variants changed with each of these alternative definitions, however FAT3 continued to be the only gene with a significant excess of cases versus controls with variants in all these tests (see Supplementary Tables S2 and S3 for altered MAF thresholds). Including synonymous variants however eliminated the case/control difference in FAT3 as well (data not shown). The protein-altering variants in FAT3 in both discovery and replication cohorts were distributed across much of the protein encoded by FAT3 (Fig. 1a).

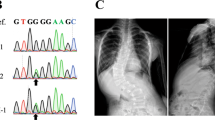
Multiplex AIS family. One family in our cohort (ID1581) consisted of three affected sisters and two unaffected parents. (a) FAT3 protein organization as annotated by NCBI is 4557 amino acids long and includes multiple functional homology domains. The positions of the 26 rare variants identified in our study among the AIS cases are labelled from 1 to 26 and are indicated by vertical arrows above the protein schema. The location of two heterozygous mutations present in this multiplex AIS family are indicated by the red boxes. (b) A simplified pedigree and segregation of the FAT3 mutations. (c) A sequence alignment with different species showing that both mutations affect an invariantly conserved amino acid sequences in FAT3 orthologues. (d) Sequence chromatograms showing those heterozygous mutations.
Whole-exome sequencing of independent AIS family
Independently of the AIS case/control cohort, we ascertained a rare multiplex family in which three sisters were affected with AIS while the parents were unaffected (Fig. 1b). Consistent with the case–control WES and targeted gene sequencing analyzes, we restricted the analysis to rare (MAF ≤ 1%), potential protein-altering SNPs or small insertions and deletions (indels). We analyzed the family WES data with different inheritance models, given the unaffected status of both parents. First, we considered a de novo mutation model in which the three sisters would share a heterozygous variant absent in the parents. Second, we considered a recessive model; either homozygous variant in the three sisters (which is heterozygous in the parents) or compound heterozygous for which the three sisters have two heterozygous variants in the same gene, each coming from one parent. Our results showed that no genes were consistent with the de novo or homozygous recessive models. However, the presence of compound heterozygous variants in FAT3 were found. Of note, the selection of candidate genes, which included the same FAT3 gene, from unbiased WES of the case–control cohort was done before we performed the family study. The two FAT3 variants found in the multiplex family are non-synonymous: p.L517S (rs139595720) and p.L4544F (rs187159256) (Fig. 1c). Both variants were confirmed by Sanger sequencing of DNA from all members of the family (Fig. 1d). The first variant was also present in four cases and one control in the replication cohort, and the second variant was present in one case in the discovery cohort.
Validation of FAT3 gene structure and identification of a novel unannotated exon
The gene model for FAT3 used by RefSeq appears to be supported mainly by long individual rodent cDNA clones in the NCBI database, whereas there are only fragmentary human cDNA clones documented in the public genome browsers. Therefore, to confirm the human FAT3 gene structure, we analyzed our in-house brain RNA-Seq data and whole-genome bisulfite sequencing (WGBS) data for one individual. Our results were consistent with the RefSeq gene model (NM_001008781.2) with two exceptions. Just upstream of the 3’ terminal exon we found evidence for two alternative exons which were either included or excluded together in various RNA-Seq reads. The two exons are also annotated by the GENCODE project website (version 24). In addition, we identified a previously uncharacterized exon located 125 kb upstream of the first annotated exon, supported by multiple individual reads splicing this sequence to the second (but first protein-coding) exon (Supplementary Fig. S1). This novel exon lies in a hypomethylated CpG island, a feature that is characteristic of active promoters (Supplementary Fig. S2). Because the 5′-most exon annotated by GENCODE (exon 2 of our gene model) begins precisely at the splice acceptor junction, we suspect that the GENCODE raw data probably included exon 1 in some junction reads, which were not aligned to the genome across exon 1 due to the very long first intron. We also profiled FAT3 expression using GTExTranscriptome Portal and observed a strong enrichment in brain and artery tissues (Supplementary Fig. S3; note that in the course of preparation of this manuscript, an additional RefSeq annotation for FAT3 has appeared, NM_001367949.1, which included an additional exon in the CpG island).
Sanger sequencing of exons 25 and 26 of FAT3
The alternative exons 25 and 26 were not captured in the replication capture sequencing because they are not annotated by RefSeq. Hence, we performed direct Sanger sequencing for these two exons in 72 cases of the first replication cohort (DNA was not available for the rest of the cases). No rare, potentially protein-altering variants were observed among the sequenced cases for either of these two (very small) exons (data not shown).
Consequences of the FAT3 rare variants
We identified in AIS patients 26 non-synonymous SNVs (25 previously reported in public databases and 1 novel) in the FAT3 gene (Table 6). Prediction of the functional consequences of the non-synonymous SNVs was performed using three different algorithms including SFIT, PolyPhen-2 and MutationTaster2. Of note, two variants were predicted as likely pathogenic by all three algorithms, 13 variants as likely pathogenic by two of the three algorithms, and one variant is a frame shift mutation (Table 6). To test whether these rare variants affect the expression of FAT3, we performed qPCR expression analysis using RNA extracted from primary osteoblasts obtained from seven scoliotic patients who had rare variants in FAT3 from the discovery and replication cohorts, and seven controls (trauma patients who did not have scoliosis and from whom we could extract osteoblasts). No statistically significant difference in averaged FAT3 expression was observed between the two groups (Supplementary Fig. S4). We did a second validation step using another independent replication cohort (replication cohort 2, Table 3) using well characterized AIS patients having reached skeletal maturity, to genotype nine FAT3 gene variants including the two variants previously identified in the multiplex family: p.L517S (rs139595720) and p.L4544F (rs187159256). Interestingly, two FAT3 variants rs139595720 (genotype A/G) and rs80293525 (genotype C/T) were enriched in scoliosis ≥ 40° (4.5% and 2.7% respectively) compared to < 40° (1.4% and 0.7%) and controls (1.2% and 0.8%). Whereas the variant rs142403035 (genotype A/G) was associated with less severe spinal deformities with a prevalence of 1.7% in scoliosis cases ≥ 40° compared to 2.1% and 4.4% in < 40° scoliosis cases and controls respectively (Table 7).
Discussion
Using WES with a combined two-stage, case/control and multiplex family approach, we discovered a new association between the FAT3 gene and AIS. Although the cohort-based gene burden test did not achieve full statistical significance after correcting for multiple gene testing, the observation of compound heterozygous variants in FAT3 in all three affected siblings in an independently ascertained multiplex AIS family (itself a very unusual occurrence), strongly supports the identification of FAT3 as an interesting candidate gene in AIS. The failure to achieve full statistical significance is likely due to the size limitation of our cohorts. It should be noted that population stratification or bias between cases and controls is unlikely since both were similarly obtained from the general Quebec school population.
Most other studies of AIS genetics have looked for individual rare variants in families23, rather than collapsing these variants by genes. POC5 and HSPG2 were initially identified from such familial studies, and only then were further investigated in independent cohorts21,22. Only two studies employed an approach similar to ours, looking at rare variant burden at the gene level. Buchan et al.20, with a two-stage approach beginning with WES of 91 severe AIS cases and a collapsing gene burden test25, followed by targeted gene resequencing in a second, much larger cohort. As in our study, no single gene achieved genome-wide significance, but the gene with the smallest p value, FBN1, was pursued in a replication cohort similar to our approach and replicated together with the related gene FBN2. In the second study, Haller et al., analyzed exome sequence data of 391 severe AIS cases and 843 controls. Again, in a genome-wide gene burden test no individual gene achieved statistical significance, therefore, they further collapsed genes according to gene ontology pathways and observed excess variation among genes implicated in the extracellular matrix, particularly collagen genes24. No collagen or fibrillin genes were among the 24 candidates in our replication cohort. Exome-wide genetic analysis are generally vulnerable to biases. However, the use of custom exon capture kits resulted in very high coverage of target gene exons, limiting false positive and negative errors.
AIS is a highly heterogeneous disease in terms of both phenotype and etiology, therefore finding a common genetic background in isolated cases is challenging. Several of the individual rare variants we observed in our case cohort were recurrent, suggestive of at least a modest founder effect. This is consistent with the elevated incidence of scoliosis in Quebec, given that our cohort was almost completely of French-Canadian ancestry. Nonetheless, there were a relatively large number of different rare variants in our cases versus matched controls. Our identification of FAT3 as a potential candidate gene with this strategy may also have depended on a very homogeneous phenotype definition in terms of sex and severity. It is also worth noting that our controls are not random population controls, but are effectively discordant since they are of individuals whose physical exam and the lack of family antecedents excludes a diagnosis of AIS or related spinal disorders. It would be interesting to revisit the total variant data sets from the previous population studies20,24 with respect to FAT3; however, those data are not available to us. More generally, our results indicate that two-stage approaches for rare variant detection in common complex diseases can yield good gene candidates for further study, even without additional criteria relying on previously known biology of the disease.
Interestingly, FAT3 is near another gene MTNR1B melatonin receptor 1B, in which a polymorphism has been associated with AIS26. The SNP in question, rs4753426, lies slightly proximal to the 5′ end of MTNR1B, and about 72 kb distal to the 3′ end of FAT3. We speculate that the observed association may be functionally related to FAT3 rather than MTNR1B function, especially as the association is strongest in Asian populations where there is typically more extended linkage disequilibrium.
FAT3 is a member of the FAT gene family comprised of FAT1, FAT2, FAT3 and FAT4, all of which are members of the cadherin super family homologous to the Drosophila gene Fat27 regulating planar cell polarity (PCP) in the Drosophila wing28. Members of the FAT cadherin subfamily have conserved structures from flies to vertebrates29. FAT3 contains multiple repeats of a cadherin repeat domain (involved in Ca+2 binding), a single laminin G domain and three EGF-like Ca+2 binding domains. The rare non-synonymous variants that we observed in our discovery and replication cohorts are distributed across much of the protein, including some in these conserved domain regions (Fig. 1A). Mutations in each of the FAT genes has been reported in many types of cancers including early T-cell precursor acute lymphoblastic leukemia30, ovarian31, and pancreatic32. It is presumed that they all represent somatic, not inherited mutations, although it is difficult to confirm this among the various sequencing studies. There is no particular known co-morbidity between AIS and such cancers. More interestingly, multiple rare variants in FAT3 were reported in families affected by the developmental disorder Hirschsprung disease33; two of the reported variants are present in our first stage discovery case cohort. As far as we know, there is no phenotypic component related to Hirschsprung in our cohorts. Although Hirschsprung disease is not obviously developmentally related to scoliosis, there are scattered reports in the literature of co-morbidity of these conditions34,35,36. Given the wide variety of developmental functions ascribed to the FAT genes, genetic associations of either common or rare variants to multiple complex disorders are plausible.
Somatic mutations in FAT3 affect cell adhesion and interaction mechanisms, beside affecting the Wnt pathway30. Members of the FAT family proteins work synergistically and antagonistically to affect many aspects of tissue morphogenesis37. It has been shown that FAT3 and FAT4 act synergistically during fusion of the vertebral arches37 through conserved interactions with components of planar polarity pathways. Fat3 knockout mice have planar polarity defects37. A recent study demonstrated that a targeted mutation in the zebrafish D. rerio ptk7 gene, whose encoded protein functions in cell communication, leads to both congenital and idiopathic scoliosis according to the timing of gene loss of function. Furthermore, mutation of the gene led to the disruption of both planar cell polarity (PCP) and Wnt/ß-catenin signaling, consistent with the contribution of these pathways to the disease38. The PCP pathways play an important role in regulating the polarity and behavior of different cells in different tissues39. Le Pabic et al.39 suggested that PCP might be involved in skeletal morphogenesis as well. They proposed a model whereby FAT3 coordinates the polarity and differentiation of chondrocytes affecting skeletal morphology. FAT3 is highly expressed in the nervous system and affects the neuronal morphology40, beside its expression in the intervertebral discs41, vertebral bone and other bone cells. As mentioned, two FAT3 variants (rs139595720 and rs187159256) are associated with scoliosis severity as demonstrated by the higher frequencies of the heterozygous genotypes in severe scoliosis (≥ 40°) compared to moderate scoliosis (< 40°) and healthy controls. According to the POLYPHEN-2 analysis, the variant rs139595720 (p.L517S) is probably damaging and await additional experiments to confirm.
We directly compared the FAT3 gene expression levels in bone cells in a subset of our patients harboring rare variants in the gene to a group of controls lacking such variants. However, these rare variants in FAT3 appeared to have no statistically significant effect on expression of the gene at least in this cell type. Somewhat unexpectedly, the statistical support for association of rare variants in FAT3 with AIS was stronger when synonymous variants were included. It has been shown that synonymous variants can affect mRNA splicing42, mRNA stability and protein expression43, and even in one case protein conformation and function44. We were not able to explore this directly due to lack of available biological materials from the particular cases in our cohort harboring such rare synonymous variants. However, the rare non-synonymous variants in our cases were not obviously clustered near exonic splice junctions.
In summary, our results implicate FAT3 as an interesting gene candidate contributing to either the occurrence or severity (or both) of AIS.
Materials and methods
Study populations
All patients with AIS were examined by orthopedic surgeons from the three pediatric centers participating in this study. A diagnosis of AIS required both history and physical examination with a minimum curvature in the coronal plane of 10°, showed by a standing postero-anterior spinal radiograph, by the Cobb method with vertebral rotation and without any known congenital or genetic disorder. Healthy children were recruited from schools in the Montreal area, and examined by a participating orthopedic surgeon. This study was approved by the institutional review boards of Sainte-Justine University Hospital, The Montreal Children’s Hospital, The Shriners Hospital for Children, and McGill University, as well as The Affluent and Montreal English School Boards. Written informed consents were given by parents or legal guardians and assents were given all minors. All methods were carried out in accordance with relevant guidelines and regulations.
Discovery AIS cohort
We selected 73 unrelated AIS cases and 70 sex- and age-matched healthy controls. All participants were of French-Canadian ancestry. Fifty of the cases were severe (Cobb angle ≥ 40°) and 23 were moderate (Cobb angle < 40°) (Table 1). Healthy controls were all scanned for spinal curvatures using a scoliometer and forward bending-test by an orthopedist surgeon. Moreover, healthy individuals with a family history of scoliosis were excluded.
Replication AIS cohort 1
Ninety-six patients of French-Canadian origin were selected for the first replication study, unrelated to each other or to the cases in the discovery cohort. Since 93% of the initial cohort were females and 68% were severe cases, the second cohort were chosen to be all females and severely affected. Thirty-six healthy French-Canadian females were recruited from Montreal schools, and an additional 60 French-Canadian females from the CARTAGENE project45,46 (Table 2).
Replication AIS cohort 2
Two-hundred fifty-eight patients of French–Canadian origin were selected for the second replication study, unrelated to each other or to the cases in the discovery and first replication cohort. One hundred forty-three healthy controls were recruited from Montreal’s schools (Table 3). All scoliosis patients reached their skeletal maturity and were divided as severe scoliosis (≥ 40°) (N = 111) or moderate scoliosis (< 40°) (N = 147).
French–Canadian multiplex family
A rare multiplex French-Canadian family with three affected sisters and healthy parents was ascertained and analyzed by WES analysis. The proband was diagnosed with AIS at the age of 13 years old with a right lumbar curve and a Cobb angle measuring 15°. Her first sister was diagnosed with AIS with a left lumbar curve measuring 23° and the second sister was also diagnosed with AIS with right thoracic curve measuring 13°.
DNA extraction
Blood was obtained by standard venipuncture. Genomic DNA was extracted from peripheral leukocytes using PureLink genomic DNA kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA).
Whole-exome sequencing of discovery cohort
Exome capture was performed using Agilent SureSelectXT Human All Exon 50 Mb v3 according to the manufacturer’s recommendations. Sequencing was done using Applied Biosystems’ SOLiD 5500xl at the Sainte-Justine University Hospital genomic platform. The average coverage of targeted sites was approximately 100X (Supplemental Information).
Targeted deep sequencing of selected genes in a replication French–Canadian AIS cohort
Twenty-four genes were chosen for resequencing in a second French–Canadian cohort. Enrichment of coding exons of these genes was done using Roche NimbleGen’s EZ Choice custom baits, with bar code multiplexing of 96 samples per lane of sequencing. Sequencing was done on an Illumina HiSeq 2000 and performed at the McGill University and Genome Quebec Innovation Centre (MUGQIC). The average coverage of targeted sites was approximately 400× (Supplemental Information). Although there was some scatter, in general there were equivalent numbers of variant calls in all genes in the control category of total common plus rare exonic, plus near intronic variants (Supplementary Table S1).
Whole-exome sequencing of a French–Canadian family
Exome capture for the multiplex family was performed using Agilent SureSelect Human All Exon 50 Mb v3 according to the manufacturer’s recommendations. Sequencing was done on an Illumina HiSeq2500 at the Sainte-Justine University Hospital genomic platform (Supplemental Information).
Sequencing data processing
The details of our bioinformatics analysis, pipeline and subsequent variant filters are shown in the Supplemental Information. Only protein coding, and near intronic regions were analyzed. Our analysis included SNPs, and small indels. The SIFT (Sorting Intolerant from Tolerant)47, PolyPhen-2 (Polymorphism Phenotyping v2)48 and MutationTaster249 algorithms were used to predict possible impact of amino acid substitutions on the structure and function of a human FAT3 protein in AIS patients harboring different FAT3 gene variants.
Sanger sequencing
Sanger sequencing was performed at the Genome Quebec Innovation Centre at McGill University. Primers were designed using the program Primer3. Sanger sequence chromatograms were analyzed using Mutation Surveyor. Exons 25 and 26 of FAT3 were not initially sequenced in the replication cohort because the custom baits used to capture the selected genes for sequencing were designed according to the RefSeq gene model, which did not include those two alternative exons. Hence, we performed Sanger sequencing for the two additional exons in 72 patients of the replication cohort. DNA of the other patients was not available. Numbering of variants in FAT3 is based on NCBI reference sequence entries NM_001008781.2 and NP_001008781.2.
Genotyping of SNPs in the FAT3 gene
Genomic DNA samples were derived from the peripheral blood of the subjects of the second replication cohort using PureLink Genomic DNA kit. Nine SNPs were genotyped in the FAT3 gene (Table 7). Multiplex PCR of the nine SNPs was performed at McGill University and Genome Quebec Innovation using standard procedures with 20 ng of template genomic DNA and HotStarTaq DNA polymerase enzyme. PCR reactions were run on the QIAxcel (Qiagen) to assess the amplification, followed by the single base extension using iPlex Thermo Sequenase. Genotypes were determined by MALDI-TOF mass-spectrometry and data were analyzed using Mass ARRAY Typer Analyser software.
Statistical analyses
In both phases of case/control analyses, we employed a collapsing gene burden test for significance testing, under the assumption that all rare, potentially protein-altering variants act in the same phenotypic direction with the same magnitude, independent of specific allele frequencies. In the few instances where an individual carried two rare variants in the same gene, these were counted as independent events generating a gene-allele burden count rather than a case count. In the first, discovery WES phase, chi-square p-values were calculated to compare the accumulation of rare variants (MAF < 0.01) in genes throughout the exome in patients versus controls, assuming a significance threshold of p = 6 × 10−6 (0.05/8150), based on the number of genes harboring at least one rare variant among either cases or controls in the WES data set. In the targeted gene phase (24 selected genes), Fisher’s exact test was used to calculate one-tailed p values for comparisons between patients and controls using GraphPad (https://www.graphpad.com/data-analysis-resource-center/#quickcalcs), with the statistical significance threshold corrected for the number of genes having a minimum number of rare variants as described under the “Results” section. When used, REVEL scores were used based on the pre-computed database; however, protein-truncating variants (stop gains, frameshift insertion/deletions), which are normally not assigned REVEL scores, were given a score of 1 for maximal predicted pathogenicity. REVEL scores are equally not assigned for intronic variants regardless whether they might affect splicing efficiency.
Validation of FAT3 gene structure
The gene model for FAT3 used by RefSeq does not appear to be supported by long individual human cDNA clones, and seems to be based on homology to several long rodent cDNAs. Therefore, to confirm the gene structure, we analyzed in-house brain RNA-Seq data and WGBS data from an unrelated individual not part of our cohorts, as well as from GENCODE public annotations. We also profiled FAT3 expression using GTExTranscriptome Portal (http://www.gtexportal.org/home/gene/FAT3).
Cell culture and RNA extraction
Primary osteoblasts were derived from bone specimens obtained intraoperatively from AIS and non-scoliotic trauma cases. Briefly, cells were grown in 10 cm2 culture dishes with Alpha Modification of Eagle’s Medium (αMEM) containing 10% fetal bovine serum (FBS) and 1% antibiotic/antimycotic at 37 °C and 5% CO2. Cells were grown until they reached confluence. Then, the cells were washed with phosphate-buffered saline (PBS 1×) twice and treated with 1 ml TRIzol, lysed and transferred to 1.5 ml tube and stored at − 80 °C. RNA was extracted using TRIzol, following the manufacturer’s instructions.
Quantitative RT-polymerase chain reaction (qRT-PCR)
Expression analyses by qRT-PCR were done in triplicate using GAPDH and PPIA (Peptidylprolyl isomerase A) as normalizing housekeeping genes (Supplemental Information).
Data availability
The known variant datasets analyzed in the current study are available in the dbSNP and gnomAD repositories. All other data generated cannot be deposited publicly as it is prohibited by our institutional review board. The corresponding author may be contacted to gain access to this data. Researches wishing to access the data will have to submit their own study’s approved protocol and consent forms for review.
References
Cheng, J. C. et al. Adolescent idiopathic scoliosis. Na. Rev. Dis. Primers 1, 15030 (2015).
Asher, M. A. & Burton, D. C. Adolescent idiopathic scoliosis: Natural history and long term treatment effects. Scoliosis Spinal Disord. 1(1), 2 (2006).
Wise, C. A. et al. Localization of susceptibility to familial idiopathic scoliosis. Spine 25(18), 2372–2380 (2000).
Wynne-Davies, R. Familial (idiopathic) scoliosis. J. Bone Joint Surg. [Br.] 50, 24–30 (1968).
Kesling, K. L. & Reinker, K. A. Scoliosis in twins: a meta-analysis of the literature and report of six cases. Spine 22(17), 2009–2014 (1997).
Ward, K. et al. Polygenic inheritance of adolescent idiopathic scoliosis: A study of extended families in Utah. Am. J. Med. Genet. A 152(5), 1178–1188 (2010).
Gorman, K. F., Julien, C., Oliazadeh, N., Tang, Q. & Moreau, A. Genetics of idiopathic scoliosis. eLS https://doi.org/10.1002/9780470015902.a0025313 (2014).
Edery, P. et al. New disease gene location and high genetic heterogeneity in idiopathic scoliosis. Eur. J. Hum. Genet. 19(8), 865–869 (2011).
Ocaka, L. et al. Assignment of two loci for autosomal dominant adolescent idiopathic scoliosis (AIS) to chromosomes 9q31.2–q34.2 and 17q25.3-qtel. J Med Genet. 45(2), 87–92 (2008).
Alden, K. J. et al. Idiopathic scoliosis: Identification of candidate regions on chromosome 19p13. Spine 31(16), 1815–1819 (2006).
Gorman, K. F., Julien, C. & Moreau, A. The genetic epidemiology of idiopathic scoliosis. Eur. Spine J. 21(10), 1905–1919 (2012).
Sharma, S. et al. A PAX1 enhancer locus is associated with susceptibility to idiopathic scoliosis in females. Nat. Commun. 6, 1–10 (2015).
Sharma, S. et al. Genome-wide association studies of adolescent idiopathic scoliosis suggest candidate susceptibility genes. Hum. Mol. Genet. 20(7), 1456–1466 (2011).
Takahashi, Y. et al. A genome-wide association study identifies common variants near LBX1 associated with adolescent idiopathic scoliosis. Nat. Genet. 43(12), 1237–1240 (2011).
Kou, I. et al. Genetic variants in GPR126 are associated with adolescent idiopathic scoliosis. Nat. Genet. 45(6), 676–679 (2013).
Ogura, Y. et al. A functional SNP in BNC2 is associated with adolescent idiopathic scoliosis. Am. J. Hum. Genet. 97(2), 337–342 (2015).
Zuk, O., Hechter, E., Sunyaev, S. R. & Lander, E. S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. 109(4), 1193–1198 (2012).
Asimit, J. & Zeggini, E. Testing for rare variant associations in complex diseases. Genome Med. 3(4), 1 (2011).
Marian, A. J. Elements of ‘missing heritability’. Curr. Opin. Cardiol. 27(3), 197–201 (2012).
Buchan, J. G. et al. Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis. Hum. Mol. Genet. 23(19), 5271–5282 (2014).
Baschal, E. E. et al. Exome sequencing identifies a rare HSPG2 variant associated with familial idiopathic scoliosis. G3 Genes Genomes Genet. 5(2), 167–174 (2015).
Patten, S. A. et al. Functional variants of POC5 identified in patients with idiopathic scoliosis. J. Clin. Investig. 125(3), 1124–1128 (2015).
Li, W. et al. AKAP2 identified as a novel gene mutated in a Chinese family with adolescent idiopathic scoliosis. J. Med. Genet. 53, 488–493 (2016).
Haller, G. et al. A polygenic burden of rare variants across extracellular matrix genes among individuals with adolescent idiopathic scoliosis. Hum. Mol. Genet. 25(1), 202–209 (2016).
Li, B. & Leal, S. M. Methods for detecting associations with rare variants for common diseases: Application to analysis of sequence data. Am. J. Hum. Genet. 83(3), 311–321 (2008).
Yang, P., Liu, H., Lin, J. & Yang, H. The association of rs4753426 polymorphism in the melatonin receptor 1B (MTNR1B) gene and susceptibility to adolescent idiopathic scoliosis: A systematic review and meta-analysis. Pain Phys. 18(5), 419–431 (2015).
Lin, D.-C. et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 46(5), 467–473 (2014).
Katoh, Y. & Katoh, M. Comparative integromics on FAT1, FAT2, FAT3 and FAT4. Int. J. Mol. Med. 18(3), 523 (2016).
Sadeqzadeh, E., Bock, C. E. & Thorne, R. F. Sleeping giants: Emerging roles for the fat cadherins in health and disease. Med. Res. Rev. 34(1), 190–221 (2014).
Neumann, M. et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood 121(23), 4749–4752 (2013).
Network, C. G. A. R. Integrated genomic analyses of ovarian carcinoma. Nature 474(7353), 609–615 (2011).
Furukawa, T. et al. Whole exome sequencing reveals recurrent mutations in BRCA2 and FAT genes in acinar cell carcinomas of the pancreas. Sci. Rep. 5, 8829 (2015).
Luzón-Toro, B. et al. Exome sequencing reveals a high genetic heterogeneity on familial Hirschsprung disease. Sci. Rep. 5, 16473 (2015).
McDonald-McGinn, D. M. et al. 22q11.2 Deletion Syndrome. Nat Rev Dis Primers 1, 15071 (2015).
Reish, O. et al. Brain anomalies, retardation of mentality and growth, ectodermal dysplasia, skeletal malformations, Hirschsprung disease, ear deformity and deafness, eye hypoplasia, cleft palate, cryptorchidism, and kidney dysplasia/hypoplasia BRESEK/BRESHECK: New X-linked syndrome?. Am. J. Med. Genet. 68(4), 386–390 (1997).
Brooks, A. S. et al. A consanguineous family with Hirschsprung disease, microcephaly, and mental retardation (Goldberg-Shprintzen syndrome). J. Med. Genet. 36(6), 485–489 (1999).
Saburi, S., Hester, I., Goodrich, L. & McNeill, H. Functional interactions between Fat family cadherins in tissue morphogenesis and planar polarity. Development 139(10), 1806–1820 (2012).
Hayes, M. et al. ptk7 mutant zebrafish models of congenital and idiopathic scoliosis implicate dysregulated Wnt signalling in disease. Nat. Commun. 5, 4777 (2014).
Le Pabic, P., Ng, C. & Schilling, T. F. Fat-Dachsous signaling coordinates cartilage differentiation and polarity during craniofacial development. PLoS Genet. 10(10), e1004726 (2014).
Deans, M. R. et al. Control of neuronal morphology by the atypical cadherin Fat3. Neuron 71(5), 820–832 (2011).
Rock, R., Schrauth, S. & Gessler, M. Expression of mouse dchs1, fjx1, and fat-j suggests conservation of the planar cell polarity pathway identified in drosophila. Dev. Dyn. 234(3), 747–755 (2005).
Cartegni, L., Chew, S. L. & Krainer, A. R. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genets. 3(4), 285–298 (2002).
Nackley, A. G. et al. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science 314(5807), 1930–1933 (2006).
Kimchi-Sarfaty, C. et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315(5811), 525–528 (2007).
Godard, B., Marshall, J. & Laberge, C. Community engagement in genetic research: Results of the first public consultation for the Quebec CARTaGENE project. Public Health Genomics 10(3), 147–158 (2007).
Awadalla, P. et al. Cohort profile of the CARTaGENE study: Quebec’s population-based biobank for public health and personalized genomics. Int. J. Epidemiol. 42, 1285–1299 (2012).
Ng, P. C. & Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31(13), 3812–3814 (2003).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7(4), 248–249 (2010).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 11(4), 361–362 (2014).
Acknowledgements
We thank the patients and families who participated in this study, and the orthopedic surgeons and nursing teams at Sainte-Justine University Hospital, The Montreal’s Children Hospital, and The Shriners Hospital for Children in Montreal. We also thank Ms. Anita Franco, project coordinator, for her technical assistance as well as Dr. Zoha Kibar (Université de Montréal, Canada) and Dr. Dawei Li (Florida Atlantic University, USA) for their critical reading of the manuscript and suggestions. We thank the genomic platform at Sainte-Justine University Hospital for their help in performing our WES experiments as well as the Genome Quebec team at McGill University for their services in performing the targeted sequencing experiment and their kind assistance in some bioinformatics analysis. Additional bioinformatics support was provided by the Bioinformatics platform of the Réseau de Médecine Génétique Appliquée (RMGA) funded by FRQS (Fonds Recherche Québec en Santé). The qRT-PCR analysis was performed at the Institute for Research in Immunology and Cancer (IRIC) at Université de Montréal. This work was supported by grants to A.M. from La Fondation Yves Cotrel de l’Institut de France, Paris, France. This work was also supported by a scholarship awarded to D.N. from the CHU Sainte‐Justine Foundation. M.E.S. is supported by Genome Canada and the Sainte-Justine University Hospital Research Center.
Author information
Authors and Affiliations
Contributions
D.N., C.J., M.E.S., and A.M. designed research; D.N., C.J., S. P. J.M., M.E.S, M.E. and W.E. performed the analyses; S. P. and J.M. contributed new analytic tools; D.N., C.J. M.E.S., M.E., W. E. and A.M. analyzed data; D.N., M.E.S., M.E., W.E. and A.M. wrote the paper; and D.N., C.J., S. P. and M.E. helped run experiments. All coauthors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
This work led to a patent application (pending) owned by Sainte-Justine University Hospital (CHU Sainte-Justine). All authors declare no other potential conflicts of interest relevant to this manuscript.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nada, D., Julien, C., Papillon-Cavanagh, S. et al. Identification of FAT3 as a new candidate gene for adolescent idiopathic scoliosis. Sci Rep 12, 12298 (2022). https://doi.org/10.1038/s41598-022-16620-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-16620-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
