
Abstract
Blepharophimosis, ptosis, epicanthus inversus syndrome (BPES) is a rare autosomal dominant genetic disease characterized by a narrowed horizontal palpehral aperture, ptosis, epicanthus inversus and telecanthus with or without premature ovarian failure. Mutations in the forkhead transcription factor 2 (FOXL2) have been shown to be responsible for BPES. We performed direct sequencing of the FOXL2 gene for molecular investigation of a Chinese family with BPES. A novel duplication mutation (c.858_868dup), resulting in a truncated protein, was detected.
Similar content being viewed by others
Blepharophimosis, ptosis, epicanthus inversus syndrome (BPES, OMIM# 110100) is a rare autosomal dominant genetic disease characterized by a narrowed horizontal palpehral aperture, ptosis, epicanthus inversus and telecanthus. BPES has been classified into two categories: type I is the complex eyelid malformation associated with premature ovarian failure (POF), and type II patients have the complex eyelid malformation only.1
Mutations in the forkhead transcription factor 2 (FOXL2) located in chromosome 3q23 region have been described as a cause of both the types of BPES. FOXL2 is a single-exon gene that is 2.7 kb in length and encodes a protein of 376 amino acids, containing a 110 amino acid DNA-binding forkhead domain (residues 54–164) and a polyalanine (poly-Ala) tract of 14 residues of unknown function (residues 221–234).2 FOXL2 is known as a transcription factor that is expressed in the developing eyelid and the granulose cells of ovarian follicles. FOXL2 has an important role in ovarian maintenance and function by regulating granulose cell proliferation and differentiation.3,4 In the present study, we report a novel duplication mutation (c.858_868dup) in a family displaying typical BPES phenotypes (Figure 1).

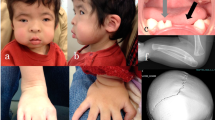
Clinical features in three patients of the family and the result of Sanger sequencing. Typical complex eyelid malformation was detected in members III:5 (a), III:10 (b) and IV:1 (c). Mutation c.858_868dup is displayed (d). Frames indicate the duplication section.
Five patients with BPES and four unaffected members of a Chinese family were recruited in the present study. BPES was inherited in a dominant autosomal pattern in the family (Figure 2). All of the patients in the family suffered from the typical complex eyelid malformation. Amblyopia occurred in the left eye of patient III:5, who had been treated twice by corrective surgeries of blepharochalasis, and occurred in both the eyes of patient IV:1. In addition, II:6 suffered from menopause at the age of 30 years, whereas II:8 suffered from irregular menstruation when she was 42 years old. Patient III:11 was approximately 19 years old and had no POF symptoms. No other symptoms were detected in any of these patients. Informed consent was obtained from the members of the family, and this study conformed to the tenets of the Declaration of Helsinki.

Pedigrees and cosegregation of mutation c.858_868dup in the family. Filled symbols represent affected individuals. The arrow indicates the proband. ‘+’ indicates individuals with a mutated allele, whereas ‘−’ indicates individuals with a normal allele.
Genomic DNA was isolated from peripheral blood by the phenol–chloroform method. The single exon and the boundary between the exon and bilateral untranslated regions in the FOXL2 gene were amplified using primer sequences designed by Primer Premier 5.0 (PremierBiosoft International, Palo Alto, CA, USA). PCR products were sequenced on an ABI 3130 Genetic Analyzer (Applied Biosystems, Carlsbad, CA, USA), and compared with a reference sequence (NCBI reference sequence: NM_023067.3). We detected a heterozygous duplication mutation (c.858_868dup) in the FOXL2 gene, which resulted in a truncated protein in the 359th codon (p.Pro290Leufs*70). This mutation was detected in affected members, and it was absent in unaffected members of the family (Figure 2).
Because the FOXL2 gene was identified as a cause of BPES,5 mutations could be detected in approximately 82% of BPES cases,6 and more than 150 mutations in the FOXL2 gene have been included in the Human Genomic Mutation Database. Although missense/nonsense mutations are primarily located in a DNA-binding forkhead domain and a poly-Ala tract, the majority of deletion, duplication and insert mutations occurred in and after the poly-Ala tract. The mutation (c.858_868dup) in our study is located in the same position after the poly-Ala tract as many other mutations, such as c.858_871dup, c.843_859dup and c.843_865dup,6 which may be a potential mutational hot spot.
Several studies have reported genotype–phenotype correlations. Among frameshift mutations, predicted protein truncations before the poly-Ala tract may lead to BPES type I, whereas predicted proteins with a poly-Ala tract expansion may be primarily BPES type II.7 However, there was no conspicuous genotype–phenotype correlations observed in our study because the typical POF did not exist in all of the female patients in the family. This may be owing to the difficulty in the diagnosis of POF because the criteria used to classify POF varied in the age of onset and the severity of ovarian dysfunction in cases with BPES.8 Genetic heterogeneity or environmental factors may be another reason for the lack of the genotype–phenotype correlation.9
In conclusion, we identified a novel mutation (c.858_868dup) in a Chinese family with BPES. This discovery further broadens the mutation spectrum of the FOXL2 gene in BPES patients, enabling clinicians and genetic counselors to offer better clinical services.
References
References
Zlotogora J, Sagi M, Cohen T . The blepharophimosis, ptosis, and epicanthus inversus syndrome: delineation of two types. Am J Hum Genet 1983; 35: 1020–1027.
Cocquet J, Pailhoux E, Jaubert F, Servel N, Xia X, Pannetier M et al. Evolution and expression of FOXL2. J Med Genet 2002; 39: 916–921.
Bentsi-Barnes IK, Kuo FT, Barlow GM, Pisarska MD . Human forkhead L2 represses key genes in granulosa cell differentiation including aromatase, P450scc, and cyclin D2. Fertil Steril 2010; 94: 353–356.
Georges A, L'Hote D, Todeschini AL, Auguste A, Legois B, Zider A et al. The transcription factor FOXL2 mobilizes estrogen signaling to maintain the identity of ovarian granulosa cells. Elife 2014; 3: e04207.
Crisponi L, Deiana M, Loi A, Chiappe F, Uda M, Amati P et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet 2001; 27: 159–166.
Beysen D, De Paepe A, De Baere E . FOXL2 mutations and genomic rearrangements in BPES. Hum Mutat 2009; 30: 158–169.
De Baere E, Dixon MJ, Small KW, Jabs EW, Leroy BP, Devriendt K et al. Spectrum of FOXL2 gene mutations in blepharophimosis-ptosis-epicanthus inversus (BPES) families demonstrates a genotype–phenotype correlation. Hum Mol Genet 2001; 10: 1591–1600.
Martinez-Aguayo A, Poggi H, Cattani A, Molina M, Romeo E, Lagos M . A novel insertion in the FOXL2 gene in a Chilean patient with blepharophimosis ptosis epicanthus inversus syndrome type I. J Pediatr Endocrinol Metab 2014; 27: 181–184.
De Baere E, Beysen D, Oley C, Lorenz B, Cocquet J, De Sutter P et al. FOXL2 and BPES: mutational hotspots, phenotypic variability, and revision of the genotype-phenotype correlation. Am J Hum Genet 2003; 72: 478–487.
Data Citations
Wu, Lingqian HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.576 (2015)
Acknowledgements
We are grateful for all of the family members and the volunteers who participated in this study. This work was supported by the National Key Technology R&D Program of China (2012BAI09B05) and the Special Fund on Scientific Research in Health Public Welfare Industry (201302001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Tan, H., Yang, P., Li, H. et al. A novel FOXL2 mutation in a Chinese family with blepharophimosis, ptosis, epicanthus inversus syndrome. Hum Genome Var 2, 15008 (2015). https://doi.org/10.1038/hgv.2015.8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.8
