
Abstract
Pulmonary acinar hypoplasia (PAH) and lacrimo-auriculo-dento-digital (LADD) syndrome have both been associated with loss-of-function variants in, or deletions of FGF10. Here we report a multi-generational family with seven members manifesting varying features of LADD syndrome, with one individual dying in early infancy of PAH. Whole genome sequencing in one family member identified a 12,158 bp deletion on chromosome 5p12 that removes two of the three exons of FGF10. Allele-specific PCR demonstrated that all affected family members, including the individual with PAH, carried the 12 kb deletion. We conclude the deletion is pathogenic and expands the mutational spectrum of FGF10 variants in LADD syndrome. The common mechanism underlying the variable clinical features of LADD syndrome is defective terminal branching of salivary and lacrimal glands and pulmonary acini, regulated by the TBX4-FGF10-FGFR2 pathway. The variable phenotypic expressivity of FGF10 haploinsufficiency from relatively benign to lethal is likely due to variation at other genetic loci.
Similar content being viewed by others


Introduction
Pulmonary acinar hypoplasia (PAH) or acinar dysplasia of the lung, is a rare neonatal disorder with a near complete fatality rate. It is characterised by absent or immature acinar structures, with no alveoli. The recurrence of PAH within families is well-recognised [1,2,3], but until recently the genetic basis of the disorder was unknown. In 2016, PAH was linked to variants in the genes TBX4 and FGFR2 [4, 5]. The protein products of these genes are components of an epithelial-mesenchymal signalling pathway that regulates the developmental branching of lungs and limbs, among other organs [4,5,6,7].
Fibroblast growth factor 10 (FGF10) is also a key component of this pathway [8]. Pathogenic variants in FGF10, along with FGFR2 and FGFR3, cause the dominantly inherited allelic disorders lacrimo-auriculo-dento-digital syndrome (LADD – OMIM 149730) and aplasia of the lacrimal and salivary glands (ALSG – OMIM 180920) [9,10,11,12]. In 2019, Karolak et al. reported five cases of PAH from four families who were heterozygous for copy-number deletions, or SNVs, in FGF10 [9].
Here, we describe an additional family with LADD syndrome caused by a novel 12 kb heterozygous deletion that encompasses the last two exons of FGF10. DNA extracted from a Guthrie card sample from a neonate who died of PAH in the early postnatal period, confirmed that she too carried the deletion.
Subjects and Methods
Clinical history
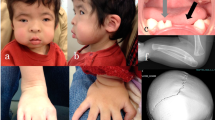
The index case (III.6, represented by an arrow on Fig. 2B) was born with hypoplasia of the left thumb (Fig. 1A) and an extra left thumb, which was removed surgically in infancy. She lost her primary teeth through severe caries at the age of 5 years and was subsequently found to have absent parotid and submandibular glands with poor salivary flow, and a bifid uvula (Fig. 1B). She has epiphora because of absent lacrimal ducts. A diagnosis of LADD syndrome was made at the age of 32 years. There was a striking three-generation history of the disorder (Fig. 2B).

A Hypoplastic thumb in the index case, an accessory thumb was removed surgically in infancy. B Bifid uvula in the index case. C Lung histology from the infant (individual IV.4) who died from pulmonary acinar hypoplasia (PAH), showing reduced density of bronchioles and absence of alveoli. D For comparison, normal lung histology from a 2-week old who died of sudden infant death syndrome (SIDS) showing normal alveolar structure. H & E stain, magnification ×250.

A Schematic representation of the NC_000005.9: g.44300489_44312646del deletion in context with the FGF10 gene. B Detailed pedigree to show degrees of affectedness in the four-generation family, the index case is marked with an arrow. Results from the allele specific PCR show that all affected individuals are heterozygous for the deletion, whereas unaffected family members, and a healthy control individual, only amplify the reference allele. No DNA was available for individual I.2.
The index case had her first child (IV.1) at the age of 27. The infant was born after a normal full-term pregnancy but, within a few minutes of birth, she developed respiratory distress and central cyanosis. In Neonatal Intensive Care she required ventilation, requiring 100% oxygen to maintain saturations above 90%. An echocardiogram on day 2 indicated persistent pulmonary hypertension of the newborn. Her respiratory status continued to deteriorate. After discussion with her parents, ventilator support was withdrawn on day 12.
The post-mortem examination showed a reduction in the number of large and small bronchi, bronchioles and an absence of alveoli, indicating a severe degree of PAH (Fig. 1C). No abnormalities were identified in any other organ. Clinical details of this case have been published previously [13]. The index case went on to have three other children, the youngest of whom had lacrimal duct hypoplasia and absent salivary glands (IV.4).
Human subjects and ethical approval
The study was conducted in accordance with the Declaration of Helsinki and the family consented to participate in a research study under approved protocols MEC/08/08/094 and 13/STH/56 (Health and Disability Ethics Committee, New Zealand). Clinical images are reproduced with consent from the family.
Genome sequencing
Genomic DNA was extracted from whole blood and genome sequencing was carried out on individual III.6, by the Kinghorn Centre for Clinical Genomics (Sydney, Australia) using the HiSeq2500 (Illumina, CA, USA) as described previously [14]. Briefly, DNA was prepared with the Illumina 30× GS (TruSeq Nano) v2.5 kit. Alignment of paired reads and variant calling was in accordance with the best practice guidelines from the Genome Analysis Tool Kit (GATK). SnpEff (v.4.3 S) and BCFtools annotate were used to annotate GVCFs. Manta (v1.6.0) was used to call large deletions and duplications using the genome sequences.
Variant confirmation
Genomic DNA was extracted as above for all but one family member. For individual IV.1, DNA was retrieved by the laboratory service at Auckland City Hospital from a heel-prick blood spot taken at birth. Allele-specific PCR was performed as follows: a common forward primer, 5′ to the deletion (on the forward strand, FGF10 is located on the reverse strand), was used together with either a reverse primer designed within the deleted region, therefore only amplifying from the non-deleted allele (product size 130 bp), or with a reverse primer located 3′ to the deleted region (product size 12,271 bp reference allele, or 112 bp deletion allele). PCR conditions were such that the reverse primer, 3′ of the deleted region, would only amplify with the common forward primer from the deletion allele (112 bp). The AmpliTaq Gold polymerase kit (Applied Biosystems, CA, USA) was used for all PCR reactions. Primer sequences are available on request.
Results
Because of the previous implication of 5p12 deletions and FGF10 variants in LADD/ASG and PAH [9, 12], we examined this region in the genome sequence of the index case. This approach identified a heterozygous 12,158 bp deletion: NC_000005.9:g.44300489_44312646del; NM_004465.1:c.326–2014_*4608del. The deletion encompasses the last two (of three) exons of FGF10 (Fig. 2A). The variant was classified pathogenic (1a) according to the ACMG/AMP guidelines [15] and has been submitted to ClinVar (SCV001548555).
The maternal grandmother (I.2), mother (II.3), two maternal aunts (II.2 and 5), and a first cousin (III.1) of the index case were also affected with LADD syndrome, as well as her son (IV.4). Her first daughter (IV.1) died in early infancy from PAH (Fig. 2B). To confirm that the deletion segregated with the phenotype, a PCR-based test was designed to specifically amplify from the reference or deleted allele as described in the methods. DNA was not available from individual I.2. DNA from individual IV.1, extracted from a heel-prick bloodspot, was highly fragmented. We were able to map the breakpoints of the deleted region, in the index case, to design PCR products to be as small as possible to increase the chances of amplification from individual IV.1. The six affected family members, from whom DNA was available, amplified both a reference allele product and a deletion allele product demonstrating that they are all heterozygous for the deletion. Two unaffected family members (IV.2 and IV.3), as well as an unrelated, healthy control individual only amplified a product from the reference allele (Fig. 2C).
The lung-enhancer region (17q23.1q23.2), previously associated with PAH in families with deletions or truncations of FGF10 or TBX4 [9], was analysed for variants in the genome sequence of index case III.6. We found eight of the non-coding variants in BCAS3 identified by Karolak et al. in individuals with PAH and copy-number variation or point mutations in TBX4 [9]. One insertion variant (rs71148400) is present on both alleles, but the expansion is two nucleotides longer on one allele. The minor allele frequency of the single nucleotide variants found varies from 0.18 to 0.01. The two common variants found in individuals, with a TBX4 mutation and PAH, by Karolak et al. (rs35827636 and rs192153557) were not found in individual III.6. The complete details of the variants found are listed in Table S1.
Discussion
Loss-of-function variants, and whole gene deletions of FGF10 have previously been implicated in LADD syndrome and PAH [9]. In the family presented here, genome sequencing identified a novel 12,158 bp deletion encompassing the last two exons of FGF10. This variant is classed as pathogenic according to the ACMG/AMP guidelines. Using a targeted, allele-specific approach, we were able to show that a further six affected individuals in the family had inherited the same deletion, including the infant affected with PAH.
This deletion removes the last two (of three) exons of FGF10. The 3′ breakpoint lies in the intron between exons one and two (Fig. 2A). The transcription initiation site of FGF10 is therefore left intact, but if a transcript were to be made from the deletion allele, it would likely be subject to nonsense-mediated decay, rendering individuals with the deletion deficient of the FGF10 gene product. Previous studies have shown that heterozygous loss-of-function of FGF10 is sufficient to cause LADD/ALSG but other inherited factors could be required for these FGF10 variants to result in PAH [9]. Entesarian et al. describe a 53 kb deletion segregating with aplasia of lacrimal and salivary glands in an extended pedigree that also removes exons 2 and 3 of FGF10. However there is no reported PAH in the family [12].
Amongst their many functions, FGF10 and its receptor are key regulators of terminal differentiation of acinar structures including the salivary and lacrimal glands [16, 17]. In the lungs, this results in the arrest of pulmonary development at 10–20 weeks’ gestational age with no alveolar formation [8, 9]. FGF10 is also involved in the development of palatal structures [18]. Two members of the family we report had a bifid uvula, a finding that has been reported previously in one patient with LADD syndrome [19]. We suggest that bifid uvula may be a feature of LADD.
How both a relatively benign condition (LADD) and a fatal one (PAH) can be caused by the same deletion or single nucleotide variant requires explanation. Karolak et al. found that individuals who develop PAH with loss-of-function TBX4 or FGF10 variants also harbour non-coding single nucleotide variants in a putative lung-enhancer region at 17q23.1q23.2, whereas individuals with the same variants and normal lungs do not [9], implicating epistatic genetic interactions. We examined the genome of the index case (III.6) and discovered eight of the non-coding variants found by Karolak et al. It is uncertain from the Karolak paper if individuals with FGF10 mutations, without PAH, were also found to have risk alleles at 17q23.1q23.2. From our data, it seems as though the eight variants found in our index case, do not predispose to PAH when inherited with a FGF10 null allele. Unfortunately, because of the fragmented state of the DNA from individual IV.1, we were unable to test the burden of variation at 17q23.1q23.2. The contribution of trans-acting factors to the manifestation of FGF10-releated LADD syndrome will be important to ascertain in future presenting families.
References
Podlech J, Richter J, Czygan P, Klein PJ, Müntefering H. Bilateral Agenesis/Aplasia of the Lungs: report of a Second Case in the Offspring of One Woman. Pediatr Pathol Lab Med. 1995;15:781–90.
Chen S, Ursell PC, Adatia I, Hislop AA, Giannikopoulos P, Hornberger LK. Prenatal diagnosis of primary pulmonary hypoplasia in fraternal twins. Ultrasound Obstet Gynecol. 2010;35:113–6.
Ramanah R, Martin A, Guigue V, Arbez-Gindre F, Piard J, Terrosi P, et al. Recurrent prenatally diagnosed isolated bilateral pulmonary agenesis. Ultrasound Obstet Gynecol. 2012;40:724–5.
Barnett CP, Nataren NJ, Klingler-Hoffmann M, Schwarz Q, Chong CE, Lee YK, et al. Ectrodactyly and Lethal Pulmonary Acinar Dysplasia Associated with Homozygous FGFR2 Mutations Identified by Exome Sequencing. Hum Mutat. 2016;37:955–63.
Szafranski P, Coban-Akdemir ZH, Rupps R, Grazioli S, Wensley D, Jhangiani SN, et al. Phenotypic expansion of TBX4 mutations to include acinar dysplasia of the lungs. Am J Med Genet A. 2016;170:2440–4.
Suhrie K, Pajor NM, Ahlfeld SK, Dawson DB, Dufendach KR, Kitzmiller JA, et al. Neonatal Lung Disease Associated with TBX4 Mutations. J Pediatr. 2019;206:286–92.
Arora R, Metzger RJ, Papaioannou VE. Multiple Roles and Interactions of Tbx4 and Tbx5 in Development of the Respiratory System. PLOS Genet. 2012;8:e1002866.
Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Dev. 1997;124:4867–78.
Karolak JA, Vincent M, Deutsch G, Gambin T, Cogné B, Pichon O, et al. Complex Compound Inheritance of Lethal Lung Developmental Disorders Due to Disruption of the TBX-FGF Pathway. Am J Hum Genet. 2019;104:213–28.
Karolak JA, Gambin T, Honey EM, Slavik T, Popek E, Stankiewicz P. A de novo 2.2 Mb recurrent 17q23.1q23.2 deletion unmasks novel putative regulatory non-coding SNVs associated with lethal lung hypoplasia and pulmonary hypertension: a case report. BMC Med Genom. 2020;13:34.
Talebi F, Ghanbari Mardasi F, Mohammadi Asl J, Bavarsad AH, Tizno S. Identification of a novel missence mutation in FGFR3 gene in an Iranian family with LADD syndrome by Next-Generation Sequencing. Int J Pediatr Otorhinolaryngol. 2017;97:192–6.
Entesarian M, Matsson H, Klar J, Bergendal B, Olson L, Arakaki R, et al. Mutations in the gene encoding fibroblast growth factor 10 are associated with aplasia of lacrimal and salivary glands. Nat Genet. 2005;37:125–8.
Odd DE, Battin Mr, Hallam L, Knight DB. Primary pulmonary hypoplasia: a case report and review of the literature. J Paediatr Child Health. 2003;39:467–9.
Mi J, Parthasarathy P, Halliday BJ, Morgan T, Dean J, Nowaczyk MJM, et al. Deletion of exon 1 in AMER1 in Osteopathia Striata with Cranial Sclerosis. Genes. 2020;11:1439.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
May AJ, Chatzeli L, Proctor GB, Tucker AS. Salivary Gland Dysplasia in Fgf10 Heterozygous Mice: a New Mouse Model of Xerostomia. Curr Mol Med. 2015;15:674–82.
Makarenkova HP, Ito M, Govindarajan V, Faber SC, Sun L, McMahon G, et al. FGF10 is an inducer and Pax6 a competence factor for lacrimal gland development. Dev. 2000;127:2563–72.
Prochazkova M, Prochazka J, Marangoni P, Klein OD. Bones, Glands, Ears and More: the Multiple Roles of FGF10 in Craniofacial Development. Front Genet. 2018;9:542–542.
Ostuni PA, Modolo M, Revelli P, Secchi A, Battista C, Tregnaghi A, et al. Lacrimo-auricolo-dento-digital syndrome mimicking primary juvenile Sjögren’s syndrome. Scand J Rheumatol. 1995;24:55–7.
Acknowledgements
We would like to thank the family for their support of this research, Dianne Webster, Stella Lai and Mark Greenslade at LabPlus, Auckland City Hospital for their help in extracting DNA from the Guthrie card sample, and Curekids for funding. This work was supported by CureKids, NZ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wade, E.M., Parthasarathy, P., Mi, J. et al. Deletion of the last two exons of FGF10 in a family with LADD syndrome and pulmonary acinar hypoplasia. Eur J Hum Genet 30, 480–484 (2022). https://doi.org/10.1038/s41431-021-00902-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00902-0
This article is cited by
-
No April fools in clinical genomics
European Journal of Human Genetics (2022)
