
Abstract
The 22q11.2 deletion syndrome is the most common microdeletion disorder, with wide phenotypic variability. To investigate variation within the non-deleted allele we performed targeted resequencing of the 22q11.2 region for 127 patients, identifying multiple deletion sizes, including two deletions with atypical breakpoints. We cataloged ~12,000 hemizygous variant positions, of which 84% were previously annotated. Within the coding regions 95 non-synonymous variants, three stop gains, and two frameshift insertions were identified, some of which we speculate could contribute to atypical phenotypes. We also catalog tolerability of 22q11 gene mutations based on related autosomal recessive disorders in man, embryonic lethality in mice, cross-species conservation and observations that some genes harbor more or less variants than expected. This extensive catalog of hemizygous variants will serve as a blueprint for future experiments to correlate 22q11DS variation with phenotype.
Similar content being viewed by others


Introduction
The 22q11.2 deletion syndrome is the most common microdeletion disorder in humans, with a prevalence of ~1 in 4,000 live births.1,2 The 22q11 deletion is for the large majority a result of non-allelic homologous recombination between low copy repeats (LCRs). The most common deletion is ~3 Mb occurring between LCRs 22-A and 22-D (~90%)3,4 and covering over 40 protein-coding genes. However, smaller deletions using other combinations of LCRs have been identified. In a study of 200 patients, 8% had LCR22-AB deletions, 2% had LCR22-AC deletions, and 2% had a deletion from downstream of LCR22-A to the typical start of LCR22-D.4 Rarer deletion sizes have also been observed.5
The clinical presentation of 22q11 patients is remarkably variable. Nevertheless, the major clinical characteristics of the syndrome are velopharyngeal abnormalities, congenital heart anomalies, a characteristic facial appearance and learning disabilities.6 In addition, patients with 22q11 deletions are at significant risk for psychiatric disorders, including up to 30% developing schizophrenia.7 The syndrome is the second most common cause of intellectual disability, accounting for ~2.4% of patients with a developmental delay.8 However, none of these features appear to be fully penetrant, and each exhibits variable expression. One potential mechanism for this phenotypic variability could reside in the variation present in the remaining allele. The hemizygous deletion could unmask recessive variants on the other allele, giving rise to a pathogenic phenotype.9,10 For example, hemizygous mutations in SNAP29 can confer cerebral dysgenesis, neuropathy, ichthyosis and keratoderma, Kousseff, or a potentially autosomal recessive form of Opitz G/BBB syndrome.11 In addition, hemizygous mutations in GP1BB were initially proposed and later demonstrated to cause Bernard–Soulier syndrome.9,12
Sorting out which variants do and do not influence phenotype in our genomes is the next great challenge for human geneticists.13 The main approach to map pathogenic variation is to link or associate variants with disease phenotypes. Mapping heterozygous variation in population studies of normal individuals is usually not sufficient to know whether or not a variant can be disease causing, as only a minority of disorders are dominant. One approach to map benign variation is via the analysis of variation present in large-scale genotyping studies of homozygous variants, including nonsense variants, and null alleles in populations of ‘normal’ individuals.14,15 Mapping the variation in the remaining alleles of genomic deletion disorders might well be another rich resource to annotate the human genome. Determining a repository of variants identified and the phenotypes accrued is a first step towards the annotation of variation in 22q11. Here we catalog the variation present in the remaining allele in 127 patients with 22q11 deletions.
Materials and methods
Sample collection
Subjects were recruited through genetics centers in Europe and the United States. Informed consent was obtained from all subjects, and the study was approved by the appropriate Institutional Review Boards.
Capture design and sequencing
Custom designed Nimblegen 12 plex arrays (NimbleGen, Inc., Madison, WI, USA) were used to capture the 22q11.2 region for 72 patients. A separate cohort of 55 patients was captured using a custom Agilent Sureselect design (Agilent Technologies, Santa Clara, CA, USA), also targeting the 22q11.2 region. Sequencing was performed on an Illumina HiSeq 2000 and FASTQ files (Illumina, Inc., San Diego, CA, USA) generated with the standard Illumina pipeline. All reads have been deposited into the ENA.16
Variant and deletion identification
Reads were aligned and variants called with an in-house pipeline using Samtools v0.1.1817, BWA v0.6.218 for alignment (option -q 15) to the reference human genome (GRCh37 as from 1000 genome project19), Picard v1.7820 to mark duplicates and GATK v2.4.921–23 for realignment, base score recalibration, and variant calling (UnifiedGenotyper, options -dcov 200 --output_mode EMIT_ALL_SITES --genotype_likelihoods_model BOTH). Variant annotation was performed with Annovar v2013Feb11.24 In addition, positions of conserved transcription factor-binding sites and ENCODE25,26 ChIP-seq regions were annotated using UCSC Table27–29 downloads. Variant positions from segmental duplications30,31 (UCSC Table download) were filtered out. The Phred-scaled likelihoods field was used to filter out likely reference position calls (reference likelihood 0, homozygous variant likelihood >70) and indeterminable calls (all likelihoods⩽70). After identifying deletion sizes (see below), heterozygous variant calls were removed from the hemizygous region by only keeping positions with a homozygous variant likelihood equal to zero and reference likelihood >70.
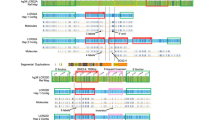
The deletion sizes, and hence the location of hemizygous variants, were determined using sequencing depths for each patient across all basepairs in chr22:18520000–22170500. These were extracted from bam files using SNIFER (E. Souche, personal communication). Basepairs in segmental duplications were flagged and all basepairs converted to coverage in 100 bp bins with custom scripts, followed by creating plots in R. Besides coverage, additional support was provided by plots of the percent of variant call types (see Figure 1 legend) in 50 kb windows.

Plots of coverage and zygosity for example LCR22-AD, LCR22-AB, LCR22-AC and LCR22-BD deletions, as well as the LCR22-A+B and LCR22-AC− patients. Blue arrows illustrate deletion sizes. For coverage, the maximum depth displayed is 600, except the low-coverage LCR22-AC− sample is set to 150, and segmental duplications are masked in gray. For zygosity plots, within 50 kb windows is indicated the percent of variants using the vcf file's Phred-scaled likelihood (PL) field as follows: bright red=confident homozygous variant (homozygous variant likelihood 0, others >70), light red=less confident homozygous variant (homozygous variant likelihood 0, heterozygous likelihood ⩽70), bright green=confident heterozygous variant (heterozygous likelihood 0, others >70), light green=less confident heterozygous variant (heterozygous likelihood 0, one of the others ⩽70).
To determine whether variant calls differed between capture platforms, sequences composed of LCR22-AD deletion variant positions were made using homozygous reference calls, homozygous variant calls, or otherwise just called N. These were used as input for MEGA5.1032 and unweighted pair group method with arithmetic mean (pairwise-deletion) was performed to create a phylogenic tree.
Variants per gene
The expected number of variants per gene was determined by first identifying non-sequenceable bases, defined as positions with only 0 or 1 reads in >75% of samples, as well as positions overlapping segmental duplications. This was done to eliminate regions that were difficult to capture or sequence. Hemizygous variants were then counted in the capture data in exonic, coding, or intronic basepairs, as defined by hg19 Refseq, as well as the number of variants between genes (i.e., intergenic regions). Variants for 1000 Genome19 data were also obtained by downloading the chromosome 22 phase 1 vcf file from the 1000 Genomes ftp site33 and similarly analyzed for the number of variants from sequenceable bases per exonic, coding, intronic or intergenic category. Plots of sequenceable length versus the number of variants were created in R and a best linear fit determined. Outliers were identified as points with Studentized residuals <−2 or >2.
The average conservation score for each gene (exonic or coding basepairs) was also determined by first downloading UCSC Comparative Genomics Conservation scores for chromosome 22 based on multiple alignments of 99 vertebrate genomes to the human genome.34,35 Similar to the above analysis, segmental duplications and regions that were difficult to capture or sequence were filtered out. The average conservation score was then calculated per exonic or coding basepair for hg19 Refseq genes in the region.
Genes were annotated for known features based on literature searches and web-based OMIM queries.36 In addition, genes were annotated for homozygous knockout phenotypes in mouse models according to the Mouse Genome Informatics WebSite.37,38
Results
Deletion size
Samples were obtained for 127 patients, targeted captures performed (55 using Agilent Sureselect capture and 72 with custom Nimblegen arrays), reads aligned and variants called. The deleted region per patient was determined based on plots of coverage and heterozygosity (Figure 1), identifying in total 8 LCR22-AB, 2 LCR22-AC, 111 LCR22-AD and 4 LCR22-BD deletions. Besides identifying common LCR-mediated breakpoints, two deletions were identified with atypical breakpoints. One deletion resembles an LCR22-AB deletion, but starts further downstream of LCR22-A (Figure 1). This is a deletion start downstream of LCR22-A similar to that reported in Shaikh et al.,4 but with the distal rearrangement in LCR22-B. Another patient was also found to have an LCR22-AC resembling deletion, but the deletion ends before LCR22-C (Figure 1). These two deletions were labeled LCR22-A+B and LCR22-AC−, respectively. Coverage plots did not indicate any large deletion events on the remaining allele.
Hemizygous variant overview
Across all patients, a total of 18,153 variant positions were initially identified in the hemizygous regions. However, upon closer scrutiny of these positions, 11.6% of Nimblegen and 24.3% of Agilent capture variants per patient were on average called heterozygous. As only a single allele is present, heterozygous variants in this region represent either mosaicisms in the original sample, or are technical artifacts. At the cost of removing potential true positives, a conservative approach was taken and heterozygous calls in the hemizygous region were removed from further analysis, leaving 11,913 variant positions (Supplementary File S1). Removal of heterozygous calls increased the percent of previously annotated variant positions from 57.8% (10,493 of 18,153 variant positions) to 84% (9,990 of 11,913 variant positions). More variants were identified per sample in DNA captured with the Nimblegen capture design as compared with the Agilent design (Supplementary Figure S1). However, when clustering hemizygous variants there was no segregation by capture technology (Supplementary Figure S2).
As larger deletions likely harbor an increased number of variants the average number of variants per patient are segregated by deletion type: 1201 LCR22-AB, 1317 LCR22-AC, 1538 LCR22-AD, 572 LCR22-BD, 587 LCR22-AC− and 835 LCR22-A+B variants (Supplementary Figure S1). As defined by Refseq, these are primarily located in introns (52%), due to the gene rich nature of the non-repetitive basepairs in the region, and intergenic regions (37%; Figure 2). Of those, 30 variants are within a conserved transcription factor-binding site and 22 variants are in ENCODE transcription factor ChIP-seq regions. A total of 1.7% of the variants (199) are located in the coding regions. A total of 44% (88) of coding variant positions are synonymous, 48% (95) non-synonymous, two insertions resulted in a frameshift and three variants resulted in a stop-gain.

Refseq based variant positions. In parenthesis is indicated the number of variant positions with corresponding annotation.
Filtering for rare variants, defined as a frequency ⩽5% in 1000 genome data and in the sequenced 22q11 patient data set, identified 2 rare stop gains, 1 rare frameshift insertion and 63 rare non-synonymous variants. The rare frameshift insertion was found in a single patient, the rare stop gains across 5 patients, and the rare non-synonymous variants across 55 patients. In total, 25 genes across 57 patients carry a rare protein-altering variant.
Toleration of gene variation and nullisomy
To determine if some genes in the region were more or less tolerant to variation, we identified genes with high and low conservation. In particular, within these 22q11 data and in 1000 genome data HIRA and PI4KA had fewer variants than expected (Figure 3), indicating these genes are more conserved and potentially less tolerant to variation. Fittingly, in the 22q11 patients HIRA had no variants affecting the open reading frame. Additional support for these genes having low tolerance to variation is that they also have high average cross-species conservation scores (Table 1).

For the 22q11 cohort of this study and 1000 genome data is plotted the number of variants found versus sequenceable length for Refseq defined genes (i.e., exonic), coding region per gene, intronic regions per gene, and intergenic regions. Blue lines are a best linear fit and black points indicate outliers.
Mutations that alter protein structure potentially affect function. Overall, protein-altering mutations were identified in 28 genes (Table 2). Seven of these genes are known for involvement in human autosomal recessive syndromes and 19 genes have been demonstrated to be embryonic lethal or result in defective phenotypes in mouse models (Table 2).
Discussion
Many genomic disorders can result in a broad phenotypic spectrum, despite having similar sized deletions or duplications. For deletions, it has been speculated that variation in the remaining allele might explain part of this phenotypic variability.9,10 Although sporadic analysis of the remaining allele has been instigated, thus far, no comprehensive analysis of a single genomic disorder has been performed to test this hypothesis. Hemizygous variant alleles present only in patients with a specific phenotype are likely to be the cause of such phenotypic differences. In addition, hemizygous alleles with variable or no phenotypic expression are likely to be benign. Considering the remarkable phenotypic variation observed for 22q11DS, we set out to explore the extent of variation in those patients, identifying a total of 11,913 hemizygous variant positions. Rare protein-altering variants are found in 57 patients and in 25 of 40 genes (Table 2).
On the basis of knockout mouse models, embryonic or neonatal lethality has been shown for eight genes in the 22q11 region (Table 2). Interestingly, here we identified non-synonymous and stop-gain mutations in six out of these eight genes. This suggests either these genes are not lethal in humans, or more likely these mutations result in partially functional proteins. Rare non-synonymous variants are found in CLDN5, CDC45, DGCR8, GNB1L and PI4KA. Four of these genes have disease associations, including all having been associated with schizophrenia.39–42 A stop-gain was identified in a single patient in TBX1. TBX1 is known to be responsible for several of the major 22q11 deletion syndrome phenotypes, including abnormal facies (conotruncal anomaly face), cardiac defects, thymic hypoplasia, velopharyngeal insufficiency with cleft palate and parathyroid dysfunction with hypocalcaemia.43 This stop-gain removes the last 19 amino acids of exon 9A of a minor isoform of the gene.44 Though TBX1 as a whole is considered essential for life, this variant proves that at least the last 19 amino acids of this isoform are not.
Patients with microdeletion syndromes are at an increased risk to manifest autosomal recessive disorders.9 Currently seven protein-coding genes within the 22q11.2 deletion region have been annotated to cause autosomal recessive conditions in humans (Table 2). Interestingly, half of these genes have protein-altering mutations in our cohort: PI4KA, PRODH, SCARF2 and SNAP29. Two non-synonymous variants (rs151146863 and chr22:21224655 C>T) and a frameshift insertion (chr22:21224770 T>TAG) were each found in SNAP29 in single previously reported individuals11 and in no additional patients in our cohort. On the basis of their atypical phenotypes (Supplementary Table S1), this demonstrates that mutations on the non-deleted chromosome can lead to unmasking of autosomal recessive conditions such as cerebral dysgenesis, neuropathy, ichthyosis and keratoderma, Kousseff, and a potentially autosomal recessive form of Opitz G/BBB syndrome.11 Notably only two genes with embryonic lethality in mice show low mutation load in this cohort: HIRA and PI4KA (Tables 1 and 2, Figure 3). Fittingly, in this cohort we did not identify any variants affecting the HIRA open reading frame. For PI4KA we did identify two rare non-synonymous positions (rs61752248 in two patients and previously unannotated chr22:21081649 G>C in a single patient with schizophrenia). The function of this gene is not well recognized, but PI4KA has previously been associated with schizophrenia, perisylvian polymicrogyria, cerebellar hypoplasia and arthrogryposis.39,47 In addition to its role in psychiatric disorders and in brain development, these results indicate PI4KA is an essential gene for human life.
Thus far 18 genes in the 22q11 region have not been associated with specific phenotypes in man or mice. Nevertheless, 11 of these genes have been identified with rare variants. Further evaluation of the patients carrying these variants may identify genotype–phenotype relationships, leading to future knowledge of gene functions.
In conclusion, we have created an extensive catalog of 22q11 hemizygous variation. These variants begin to provide insight into phenotypic contributions for the genes in the region, as well as tolerability of 22q11 gene variation and nullisomy. This catalog will serve as a blueprint for future experiments to correlate 22q11DS variation with phenotype and provides as an example for the challenges linking diverse phenotypes with large numbers of variants as we move from targeted to genome-wide sequencing.
References
Devriendt K, Fryns JP, Mortier G, van Thienen MN, Keymolen K . The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet 1998; 35: 789–790.
Oskarsdóttir S, Vujic M, Fasth A . Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch Dis Child 2004; 89: 148–151.
Carlson C, Sirotkin H, Pandita R, Goldberg R, McKie J, Wadey R et al. Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am J Hum Genet 1997; 61: 620–629.
Shaikh TH, Kurahashi H, Saitta SC, O'Hare AM, Hu P, Roe BA et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet 2000; 9: 489–501.
Weksberg R, Stachon AC, Squire JA, Moldovan L, Bayani J, Meyn S et al. Molecular characterization of deletion breakpoints in adults with 22q11 deletion syndrome. Hum Genet 2007; 120: 837–845.
Swillen A, Vogels A, Devriendt K, Fryns JP . Chromosome 22q11 deletion syndrome: update and review of the clinical features, cognitive-behavioral spectrum, and psychiatric complications. Am J Med Genet 2000; 97: 128–135.
Bassett AS, Chow EW . Schizophrenia and 22q11.2 deletion syndrome. Curr Psychiatry Rep 2008; 10: 148–157.
Rauch A, Hoyer J, Guth S, Zweier C, Kraus C, Becker C et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A 2006; 140: 2063–2074.
Budarf ML, Konkle BA, Ludlow LB, Michaud D, Li M, Yamashiro DJ et al. Identification of a patient with Bernard-Soulier syndrome and a deletion in the DiGeorge/velo-cardio-facial chromosomal region in 22q11.2. Hum Mol Genet 1995; 4: 763–766.
Hochstenbach R, Poot M, Nijman IJ, Renkens I, Duran KJ, Van't Slot R et al. Discovery of variants unmasked by hemizygous deletions. Eur J Hum Genet 2012; 20: 748–753.
McDonald-McGinn DM, Fahiminiya S, Revil T, Nowakowska BA, Suhl J, Bailey A et al. Hemizygous mutations in SNAP29 unmask autosomal recessive conditions and contribute to atypical findings in patients with 22q11.2DS. J Med Genet 2013; 50: 80–90.
Kunishima S, Imai T, Kobayashi R, Kato M, Ogawa S, Saito H . Bernard-Soulier syndrome caused by a hemizygous GPIbβ mutation and 22q11.2 deletion. Pediatr Int 2013; 55: 434–437.
Cutting GR . Annotating DNA variants is the next major goal for human genetics. Am J Hum Genet 2014; 94: 5–10.
Yngvadottir B, Xue Y, Searle S, Hunt S, Delgado M, Morrison J et al. A genome-wide survey of the prevalence and evolutionary forces acting on human nonsense SNPs. Am J Hum Genet 2009; 84: 224–234.
MacArthur DG, Tyler-Smith C . Loss-of-function variants in the genomes of healthy humans. Hum Mol Genet 2010; 19: R125–R130.
Leinonen R, Akhtar R, Birney E, Bower L, Cerdeno-Tárraga A, Cheng Y et al. The European Nucleotide Archive. Nucleic Acids Res 2011; 39: D28–D31.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
DePristo M, Banks E, Poplin R, Garimella K, Maguire J, Hartl C et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genet 2011; 43: 491–498.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013; 11: 11.10.1–11.10.33.
Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489: 57–74.
Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 2013; 41: D56–D63.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM et al. The human genome browser at UCSC. Genome Res 2002; 12: 996–1006.
Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 2004; 32: D493–D496.
Karolchik D, Barber GP, Casper J, Clawson H, Cline MS, Diekhans M et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res 2014; 42: D764–D770.
Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S et al. Recent segmental duplications in the human genome. Science 2002; 297: 1003–1007.
Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE . Segmental duplications: organization and impact within the current human genome project assembly. Genome Res 2001; 11: 1005–1017.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S . MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011; 28: 2731–2739.
1000 Genome ftp site: phase 1 vcf files. http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase1/analysis_results/integrated_call_sets/.
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A . Detection of non-neutral substitution rates on mammalian phylogenies. Genome Res 2010; 20: 110–121.
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005; 15: 1034–1050.
Online Mendelian Inheritance in Man, McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD). http://omim.org/. Accessed 26 February 2015.
Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE . Mouse Genome Database Group. The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res 2014; 42: D810–D817.
The Mouse Genome Informatics Web Site. http://www.informatics.jax.org/. Accessed 9 February 2015.
Jungerius BJ, Hoogendoorn ML, Bakker SC, Van't Slot R, Bardoel AF, Ophoff RA et al. An association screen of myelin-related genes implicates the chromosome 22q11 PIK4CA gene in schizophrenia. Mol Psychiatry 2008; 13: 1060–1068.
Williams NM, Glaser B, Norton N, Williams H, Pierce T, Moskvina V et al. Strong evidence that GNB1L is associated with schizophrenia. Hum Mol Genet 2008; 17: 555–566.
Sun ZY, Wei J, Xie L, Shen Y, Liu SZ, Ju GZ et al. The CLDN5 locus may be involved in the vulnerability to schizophrenia. Eur Psychiatry 2004; 19: 354–357.
Zhou Y, Wang J, Lu X, Song X, Ye Y, Zhou J et al. Evaluation of six SNPs of MicroRNA machinery genes and risk of schizophrenia. J Mol Neurosci 2013; 49: 594–599.
Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S et al. Role of TBX1 in human del22q11.2 syndrome. Lancet 2003; 362: 1366–1373.
Gong W, Gottlieb S, Collins J, Blescia A, Dietz H, Goldmuntz E et al. Mutation analysis of TBX1 in non-deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J Med Genet 2001; 38: E45.
Racedo SE, McDonald-McGinn DM, Chung JH, Goldmuntz E, Zackai E, Emanuel BS et al. Mouse and human CRKL is dosage sensitive for cardiac outflow tract formation. Am J Hum Genet 2015; 96: 235–244.
Yamamoto GL, Aguena M, Gos M, Hung C, Pilch J, Fahiminiya S et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet 2015; 52: 413–421.
Pagnamenta AT, Howard MF, Wisniewski E, Popitsch N, Knight SJ, Keays DA et al. Germline recessive mutations in PI4KA are associated with perisylvian polymicrogyria, cerebellar hypoplasia and arthrogryposis. Hum Mol Genet 2015; 24: 3732–3741.
Acknowledgements
We wish to thank Cédric Le Caignec and Sonia Bouquillon for providing samples and Erika Souche for assistance in running SNIFER. MSH is a postdoctoral fellow at KU Leuven supported by F+/12/037. BAN is funded by a Grant from the Foundation for Polish Science (Homing Plus/2012-5/9). This study was supported by National Institutes of Health grants MH 87626 (REG), MH 101720 (STW), MH 87636 (DMM-M and BSE), MH 101719-01 (DMM-M), and HD 070454 (DMM-M). This work was also made possible by grants from the ‘Fondation Jerome Lejeune’ and from the University of Leuven (KU Leuven), SymBioSys (PFV/10/016) and GOA/12/015 to JRV.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
DMM-M has given presentations on 22q11.2 for Natera. The authors declare no additional conflict of interests exist.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website (http://www.nature.com/hgv)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hestand, M., Nowakowska, B., Vergaelen, E. et al. A catalog of hemizygous variation in 127 22q11 deletion patients. Hum Genome Var 3, 15065 (2016). https://doi.org/10.1038/hgv.2015.65
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.65
