
Abstract
Connectivity is an important component of metapopulation dynamics in marine systems and can influence population persistence, migration rates and conservation decisions associated with Marine Protected Areas (MPAs). In this study, we compared the genetic diversity, gene flow and population structure of two octocoral species, Eunicella verrucosa and Alcyonium digitatum, in the northeast Atlantic (ranging from the northwest of Ireland and the southern North Sea, to southern Portugal), using two panels of 13 and 8 microsatellite loci, respectively. Our results identified regional genetic structure in E. verrucosa partitioned between populations from southern Portugal, northwest Ireland and Britain/France; subsequent hierarchical analysis of population structure also indicated reduced gene flow between southwest Britain and northwest France. However, over a similar geographical area, A. digitatum showed little evidence of population structure, suggesting high gene flow and/or a large effective population size; indeed, the only significant genetic differentiation detected in A. digitatum occurred between North Sea samples and those from the English Channel/northeast Atlantic. In both species the vast majority of gene flow originated from sample sites within regions, with populations in southwest Britain being the predominant source of contemporary exogenous genetic variants for the populations studied. Overall, historical patterns of gene flow appeared more complex, though again southwest Britain appeared to be an important source of genetic variation for both species. Our findings have major conservation implications, particularly for E. verrucosa, a protected species in UK waters and listed by the IUCN as ‘Vulnerable’, and for the designation and management of European MPAs.
Similar content being viewed by others

Introduction
Population connectivity has emerged as a key factor in the sustainable management of marine resources (Fogarty and Botsford, 2007; Da Silva et al., 2014), in tracking invasive species (Pérez-Portela et al., 2012), in monitoring the effects of climate change (Munday et al., 2009; Gerber et al., 2014), and in designating networks of protected areas (Jones et al., 2007; Marti-Puig et al., 2013). For most benthic marine organisms, connectivity is typically defined by dispersal during early life stages and is intimately associated with oceanic currents and topographical features (Cowen et al., 2007). However, connectivity can vary across marine taxa, even between closely related species over similar spatial scales (Bargelloni et al., 2003, 2005; Charrier et al., 2006; Kool et al., 2013) and population structure can be determined by the extent of dispersal from distant vs local sources, resulting in fully ‘open’ (panmictic) to fully ‘closed’ (isolated) populations (see Cowen and Sponaugle, 2009 and references therein). Perhaps most importantly from an applied perspective, population structure and gene flow can be used as a proxy for understanding population connectivity (Hedgecock et al., 2007; Lowe and Allendorf, 2010; Kool et al., 2013).
Advances in our knowledge of marine population connectivity are fundamental for the strategic allocation of available resources in a way that maximises protection of marine biodiversity (Kool et al., 2013). Moreover, the global extent of protected areas is unlikely to mitigate the current rate of marine and terrestrial biodiversity loss (Mora and Sale, 2011). Among the 15 European countries that have signed the Oslo/Paris (OSPAR) Convention (for the protection of the marine environment of the northeast Atlantic), there is a requirement to establish an ‘ecologically coherent’ network of Marine Protected Areas (MPAs), which collectively aims to deliver more benefits to biodiversity than single, unrelated MPAs (OSPAR Convention, 2013). As connectivity is a key feature of an MPA network, it is important that empirical estimates of population connectivity are considered during the designation or review stages of a network (Jones et al., 2007). For example, guidelines for incorporating connectivity into designing networks of marine reserves are available for coral reefs and are likely to be useful for the management and protection of these systems (Almany et al., 2009; McCook et al., 2009). Several analyses of connectivity in established networks have also identified deficiencies that may reduce the efficacy of a network. For example, Puckett et al. (2014) modelled dispersal of the eastern oyster (Crassostrea virginica) on the Atlantic coast of North Carolina and showed that if marine reserves were too small – relative to the mean dispersal distance of the oyster– local retention of larvae was reduced; likewise, if reserves were spaced too far apart, connectivity became limited. While early landmark studies of genetic connectivity in the marine environment (for example, Palumbi, 2003) focused largely on gene flow, barriers to gene flow and isolation by distance (IBD), more recent studies (for example, Arizmendi-Mejía et al., 2015; Gagnaire et al., 2015) have further refined our understanding of drivers of marine genetic connectivity and have demonstrated the importance of additional factors in driving or disrupting genetic connectivity, for example, effective population size and genetic drift. Overall, such findings suggest a greater understanding of population structure and connectivity is required to optimise the conservation of marine biodiversity and to maximise the efficacy of such networks (for example, OSPAR Commission, 2006; Jones et al., 2007)
Currently, and until such time as a robust understanding of the functioning of networks of MPAs is achieved, individual MPAs are typically designated based on the presence of rare or protected species or guilds of species; for example, in the waters of southwest Britain, the presence of Eunicella verrucosa (the pink sea fan) is often listed as a factor in the designation of an area as a Marine Conservation Zone (MCZ). However, population genetic studies of octocorals across this area, and the northeast Atlantic in general, are limited. Previous research in this region has had either a phylogenetic (for example, McFadden and Hutchinson, 2004) or phylogeographic focus (for example, Herrera et al., 2012), while existing connectivity research on this subclass in the region has assessed the genetic diversity and structure of primarily Mediterranean species, for example, Corallium rubrum (Costantini et al., 2007; Ledoux et al., 2010; Aurelle et al., 2011), Eunicella singularis (Costantini et al., 2016), Eunicella cavolini (Masmoudi et al., 2016) and Paramuricea clavata (Mokhtar-Jamai et al., 2011; Arizmendi-Mejía et al., 2015). As a result, genetic diversity and connectivity in this group remains understudied.
Eunicella verrucosa is an IUCN red-listed octocoral. It can be found from Angola to western Ireland, but its range in the British Isles is limited to southwest England, southwest Wales, and southern and western Ireland (Hayward and Ryland, 1995). In Britain, it is considered rare due to its limited distribution beyond the southwest (Hiscock et al., 2010), although where it is found it can be relatively abundant and may form ‘forests’. Colonies are generally found inhabiting rocky substrates, at depths of 10–150 m, in areas of high turbidity with moderate to high current flow. E. verrucosa has an important role for the functional ecology of sublittoral ecosystems in which it occurs; it provides structural complexity and habitat for numerous epifauna and, as such, may be considered to be an ecosystem engineer (Hall-Spencer et al., 2007; Pikesley et al., 2016). Colonies are also vulnerable to trawling activity and, as a result, the designation of several MPAs across Britain includes E. verrucosa as a specific factor (a ‘protected feature’) in their designation.
Alcyonium digitatum (‘dead man’s fingers’) has a ubiquitous presence along rocky upper and circalittoral zones, typically to a depth of 200 m, and it can be found around most British and Irish coasts (Hayward and Ryland, 1995); it is represented in several MPAs across the UK network. It is widely distributed across the North Atlantic, ranging from Portugal to Norway, to eastern Canada, south to Cape Hatteras in the USA (Hartnoll, 1975; Watling and Auster, 2005). It is not a protected species, however, it is locally depleted in some areas by benthic trawling (Hinz et al., 2011). Both species are thought to be lecithotrophic, gonochoristic (separate sexes) and broadcast spawners, with limited reports of hermaphroditism in A. digitatum; asexual reproduction may also be possible in E. verrucosa as genets can proliferate via fragmentation (Hartnoll, 1975; McFadden et al., 2001; Munro, 2004). Alcyonium digitatum releases gametes in winter (December–January) and pelagic larvae can survive up to 14 weeks and beyond (Hartnoll, 1975). Spawning of E. verrucosa occurs towards the end of summer (August–September), though its pelagic larval duration is unknown (Munro, 2004). Studying patterns in genetic connectivity and assessing genetic diversity offers an alternative approach by which to infer the dispersal capabilities of these species.
In this study, two panels of microsatellites (Holland et al., 2013a, 2013b) were used to assess the population structure and genetic connectivity of E. verrucosa and A. digitatum around the British Isles and northeast Atlantic. Specifically, we addressed the following questions: (i) what is the genetic diversity of each species and is it uniform across the sampling range; (ii) do both of these species show population genetic structure indicative of departures from panmixia; and (iii) what are the levels of gene flow and effective population size for each species? Finally, we consider the conservation and potential management implications of our findings for these species, both in terms of connectivity between existing MPAs and with regard to the designation of future MPAs.
Materials and methods
Study sites and sampling
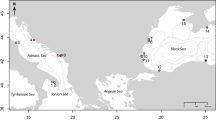
Samples of E. verrucosa (N=922) were collected from 27 sites ranging from southern Portugal to northwest Ireland, including sites around Brittany in northwest France, Lyme Bay in southern England and southwest Wales (Table 1 and Figure 1). The area sampled represents much of the northern range of the species. Samples of A. digitatum (N=655) were collected from 20 sites across a similar geographic area (with the exception of southern Portugal, where the species was not found); samples from two additional sites in the North Sea (Table 2 and Figure 1) were also included. The area sampled represents much of the southern range of A. digitatum in Europe. Samples of both species were collected between 2007 and 2012. The majority of samples were collected by SCUBA at depths between 10 and 35 m; additional samples of A. digitatum were collected by trawling (CEFAS scientific trawl, Lowestoft, UK). Colonies of E. verrucosa were sampled by removing a 3 cm terminal branch using sea-snips. This species is protected in UK waters, and all UK sampling complied with licenses granted by Natural England and the Marine Management Organisation (see Acknowledgements). Colonies of A. digitatum were sampled by removing a 1 cm3 section of tissue from a terminal thumb-like ‘branch’ using sea-snips. After removal, individual colonies were placed into mesh bags, brought to the surface, and quickly immersed in 95–100% ethanol for storage. In both species, samples were taken from individual colonies spaced at least 1 m apart to avoid sampling clones; previous studies of hard corals have identified potential clones at spatial scales up to 5 m apart (for example, Goffredo et al., 2009; Foster et al., 2012). This issue was also addressed after genotyping by identifying and excluding any duplicate genotypes occurring in the same population.

Map of the sites sampled in the northeast Atlantic. Pink circles represent sites where only Eunicella verrucosa were collected and blue circles represent where only Alcyonium digitatum were collected. Circles containing both colours represent sites in which both E. verrucosa and A. digitatum were collected. See Table 1 for details on population codes, sample size and latitude and longitude.
DNA extraction and microsatellite genotyping
Total genomic DNA was extracted from ~10 to 20 polyps using a WizardR SV Genomics DNA Purification System kit (Promega, Southampton, UK) following the manufacturer’s protocol. Polyps were removed from colonies using forceps, or by using a scalpel to shave a portion of ~1 cm2 surface tissue from A. digitatum or 1–2 cm of coenenchymal tissue (excluding the gorgonin axis) from E. verrucosa. Microsatellites were amplified for both species and alleles were scored using GENEMAPPER v3.7 (Applied Biosystems, Paisley, UK). Full details of DNA extraction and microsatellite amplification conditions and multiplexing are given in a primer note for each species: E. verrucosa (Holland et al., 2013a) and A. digitatum (Holland et al., 2013b).
Data screening and quality assessment
Duplicate genotypes were identified in CERVUS v3.0.3 (Kalinowski et al., 2007) and were removed from further analyses. The presence of possible null alleles, allele scoring errors due to stuttering and large allele dropout was evaluated using MICRO-CHECKER v2.2.3 (Van Oosterhout et al., 2004). Linkage disequilibrium and deviation from Hardy–Weinberg equilibrium (HWE) were tested in GENEPOP v4.2 (Rousset, 2008) using default parameters and the false discovery rate was used to detect type-1 errors (Storey and Tibshirani, 2003). To identify candidate markers under selection or linked with markers under selection, loci were screened using two different FST outlier detection methods in Lositan (Antao et al., 2008) and Arlequin v3.5.2 (Excoffier and Lischer, 2010). Lositan assumes an island model and runs were conducted using the infinite alleles model. Parameters were set to 50 000 simulations, a 99% confidence interval and a false discovery rate of 0.1, with the neutral and forced mean FST enforced. In Arlequin, 50 000 simulations were run with 100 demes simulated per group and 10 simulated groups under the hierarchical island model. Samples were grouped by geographical region (E. verrucosa: Portugal, France, Ireland, Britain; A. digitatum: France, Ireland, Britain, North Sea) and results were considered significant if the P-value was <0.010.
Genetic variation
Expected heterozygosity (Hexp) and the inbreeding coefficient (FIS) for each population were estimated using the diveRsity package (Kennan et al., 2013) in R (R Development Core Team, 2016). The divBasic function was used and FIS significance was assessed using 95% confidence intervals using 1000 bootstrap replicates; the significance level for multiple comparisons was corrected using a Bonferroni correction (Dunn, 1961), which had the effect of slightly widening each interval. Allelic richness (Ar) and private allelic richness (PAr) were calculated in HP-RARE v1.1 (Kalinowski, 2005) using a rarefaction method, which accounts for variation in sample size (each sample included a minimum of eight loci).
Population structure
Population differentiation was analysed using pairwise FST (Weir and Cockerham, 1984) and G”ST (Meirmans and Hedrick, 2011) measures using the diffCalc function in diveRsity, and significance was assessed as for FIS. To search for genetic structuring, a principal coordinates analysis (PCoA) was performed using a matrix of codominant genotypic genetic distances in GenALEx v6.5 (Peakall and Smouse, 2012). An analysis of molecular variance (AMOVA) was performed using Arlequin (10 000 permutations) to test for differentiation amongst geographical regions. Population structure was also analysed using a Bayesian clustering method: STRUCTURE v2.3.4 (Pritchard et al., 2000), using a burn-in of 104 and 106 repetitions. An admixture ancestry model using population IDs as priors and correlated allele frequencies was chosen. To determine the number of populations (K), the delta K statistic (Evanno et al., 2005) and the mean value of L(K) were examined in the POPHELPER R package (Francis, 2017). Ten replicate runs were aligned and merged in POPHELPER using CLUMPP and graphics were generated using the merged data. Initial runs for both species using K values 1–10 showed a very low likelihood for K values 6–10, therefore, subsequent runs included only K values of 1–5. A Mantel test was implemented in GenALEx to test whether any observed genetic structure was a product of IBD. Genetic distances were supplied as FST/(1- FST) matrices and were compared with the logarithm of geographic distances (km). Negative FST values were converted to zero for this analysis. Geographical distances were estimated in Google Earth by measuring the shortest in-water distance between sites in a straight line or by calculating the shortest distance following coastlines.
Gene flow and effective population size
Contemporary (within the last few generations) and historical gene flow was estimated using two methods. Contemporary gene flow was analysed using BayesAss v3.0.4 (Wilson and Rannala, 2003), which estimates the fraction of immigrants in a population using Bayesian inference. Three runs were performed using 107 iterations, a burn-in of 106 and a sampling interval of 100, and an average of the gene flow estimates was calculated. The mixing parameters DeltaA, DeltaF and DeltaM were set to 0.10, 0.20 and 0.05 for E. verrucosa, and 0.30, 0.50 and 0.10 for A. digitatum, respectively. Convergence of the chains was validated using Tracer v1.6 (Rambaut et al., 2014). Historical gene flow was calculated using the mutation-scaled migration rate M (m/μ; where m is the immigration rate per generation) and the population parameter theta (4Ne*μ) in Migrate-n v3.6 (Beerli and Felsenstein, 2001). Migrate-n is a coalescence-based program that has the benefit of providing values of immigration and emigration for each population and is therefore useful in scenarios of asymmetrical migration. A Brownian motion model was used and assumed a migration matrix with variable theta and estimated mutation rates for loci based on the data. A Bayesian likelihood strategy was initially run with default parameters to obtain start parameter estimates for theta and M. These parameters were supplied to the program in subsequent runs and the number of recorded steps was increased to 50 000. Prior uniform distributions for theta and M were set to min=0, max=100 and delta=10, and min=0, max=1000 and delta=100, respectively. To evaluate convergence of the chains, the effective sample size (>1000) and the shape of the histograms in the output files were examined.
Migrate-n was also used to calculate the mutation-scaled effective population size (Ne). This was calculated from the optimum value of theta using the equation Ne=theta/4 μ, assuming a microsatellite mutation rate (μ) of 10−4 per generation, as used in a previous study of a Mediterranean cup coral (Casado-Amezú et al., 2012). Contemporary Ne was estimated using LDNE v1.31 (Waples and Do, 2008). The program was run assuming a model of random mating and the allowed frequency of observed alleles was set to 0.050.
Results
Data screening and quality assessment
For E. verrucosa, based on evidence of null alleles and significant deviation from HWE, one locus (Ever009) was omitted from the original microsatellite panel of Holland et al. (2013a). Five other loci also showed some deviation from HWE, however, departures from HWE occurred in only a few populations and these loci were retained. Similarly, linkage disequilibrium was detected in five populations, but each population showed different pairs of potentially linked loci. With no obvious trend in the pattern of linkage disequilibrium observed, this inconsistency was likely due to site-specific biological processes which we were not able to investigate further within this study; consequently, no loci were discarded on the basis of linkage disequilibrium and 13 were used for subsequent analyses. For A. digitatum, three loci (Adig003, Adig004 and Adig010) were discarded from the original microsatellite panel of Holland et al. (2013b) based on the presence of null alleles and significant deviations from HWE. Some evidence of linkage disequilibrium was also detected, but was minimal across populations and no further loci were omitted; eight loci were used for subsequent analyses.
A relatively low number of duplicate genotypes were identified in both species. In E. verrucosa, 17 individuals with duplicate genotypes were identified in nine samples (Table 1), while in A. digitatum, seven individuals with duplicate genotypes were identified in five samples (Table 2). Duplicates were removed from further analyses. The spread of duplicates across sites did not show any obvious pattern in either species, with the exception of a small sample of E. verrucosa (Cam) from north Cornwall, in which four duplicate individuals (across three genotypes) were identified out of a sample of only 11 individuals successfully genotyped.
E. verrucosa samples were monomorphic at several loci, but this was not consistent in all populations at the same locus. In comparison, A. digitatum was monomorphic at only one locus (Adig007) in three populations. For E. verrucosa, two loci (Ever013 and Ever014) were identified as outliers under the island model and one (Ever013) under the hierarchical island model (Supplementary Appendix 1). Accordingly, as both methods identified Ever013 as an outlier under positive selection, analyses of population structure, gene flow and effective population size excluded this locus; STRUCTURE, PCoA and BayesAss analyses were conducted using 13 loci as the assumptions of these methods are not violated by the inclusion of loci under selection (Pritchard et al., 2000; Wilson and Rannala, 2003). One outlier locus (Adig006) was identified for A. digitatum by the island model, but not by the hierarchical island model (Supplementary Appendix 1); accordingly, eight loci were retained.
Genetic variation
After removal of duplicate genotypes, genotypes of 905 individual specimens of E. verrucosa from 27 sites were analysed at 13 loci. For A. digitatum, genotypes of 648 individual specimens from 20 sites were analysed at eight loci. For E. verrucosa, measures of Hexp ranged from 0.367 (Black Rock) to 0.459 (Hand Deeps) and were generally consistent within regions, with minor differences between some regions (Table 1). A similar pattern was observed for Ar, which ranged from 2.38 (Black Rock) to 2.79 (The Heroine Wreck); overall, both measures were slightly lower in the samples from Ireland. For A. digitatum, Hexp and Ar measures were also relatively uniform within and between regions (Table 2) and were consistently higher than for E. verrucosa; Hexp measures ranged from 0.594 (The Lucy Wreck) to 0.668 (Norfolk) and Ar ranged from 3.99 (Laonegued Taer) to 4.34 (Roscoff2). Private allelic richness (PAr) was also consistently higher for A. digitatum than for E. verrucosa; values for A. digitatum ranged from 0.075 (The Lucy Wreck) to 0.193 (Roscoff1), while values for E. verrucosa were between 0.001 (Camel Estuary) to 0.059 (Laonegued Taer). The majority of FIS values for both species were positive; overall, however, few were significant, though generally at least one site in each region showed a significant positive FIS coefficient (Tables 1 and 2). This finding suggested a deficiency of heterozygotes at some sites; for E. verrucosa, this was most apparent in several populations from Portugal, while both species showed significant, positive FIS values at Roscoff2. The broader implications of these findings (inbreeding and/or a Wahlund effect caused by the inadvertent combining of data from separate populations) are discussed below. A small sample of E. verrucosa from the Camel Estuary (Cam) had a significantly negative FIS, indicating an excess of heterozygotes at this site.
Population structure
Global FST and G”ST measures across all populations of E. verrucosa were 0.012 and 0.023, respectively (Supplementary Appendix 2). In comparison, global FST and G”ST measures for all populations of A. digitatum were lower (0.003 and 0.011, respectively) (Supplementary Appendix 2). For both species, global values were significantly different from zero.
For E. verrucosa, the largest significant pairwise FST (0.059) value was observed between Faro and the Camel Estuary, while the highest significant pairwise G”ST (0.089) value observed was also between populations from Portugal and southwest Britain: Portamao2 and the Heroine Wreck (Supplementary Appendix 2). In contrast, the highest significant pairwise FST and G”ST values (0.020 and 0.058, respectively) for A. digitatum were between populations from southwest Britain and the North Sea: Trenemene Reef and Norfolk (Supplementary Appendix 2). For both species, both pairwise measures were typically low and non-significant within regions and between populations from Britain and France. For A. digitatum, only pairwise comparisons with North Sea populations were significant. However, for E. verrucosa, many pairwise comparisons between Portugal populations and populations from Britain, Ireland and France were significantly different from zero.
The PCoA suggested regional structure in E. verrucosa (Figure 2a), with evidence for three clusters: Portugal, Ireland, and populations from Britain and France. In contrast, little evidence of regional structure was apparent in A. digitatum (Figure 2b). There was some evidence for the isolation of the North Sea and UB74 Wreck populations of A. digitatum; however, genetic structure did not appear wholly concordant with geography, as the North Sea populations did not group together.

PCoA for Eunicella verrucosa (a) and Alcyonium digitatum (b). Colours correspond to regions: Britain (red), France (blue), Ireland (green), Portugal (orange), the North Sea (purple).
For the AMOVA, populations were grouped by geographical region for each species: Portugal, France, Ireland and Britain (E. verrucosa) and France, Ireland, Britain, and the North Sea (A. digitatum). In both species, global tests revealed that the majority of variation was explained by variation within populations (Supplementary Appendix 3). For E. verrucosa, a small but highly significant amount of variation was explained by differences between the geographical regions (FCT=0.016, P<0.001). Similarly, a significant (but much smaller) amount of variation was explained by differences between regions for A. digitatum (FCT=0.001, P=0.049).
For E. verrucosa, both the mean L(K) and delta K statistics indicated K=3 as the most probable number of discrete populations within the data set (Supplementary Appendix 4). STRUCTURE analysis (Figure 3a) identified essentially the same groupings as observed in the PCoA (Figure 2a), but also indicated that all E. verrucosa colonies from France (and a few from Britain) shared some allelic similarities with E. verrucosa from Portugal. To explore potentially finer-scale population structure (<500 km distance between sites) in populations from Britain and France, a hierarchical STRUCTURE analysis was conducted using data from only these regions. The most likely number of populations was identified as K=2 (Supplementary Appendix 4), which revealed moderate structure partitioned between E. verrucosa populations from Britain and those from France, with some evidence of allelic variants more typical of E. verrucosa from France occurring in samples from Britain (Figure 3b). In contrast, for A. digitatum, the mean L(K) suggested panmixia (K=1; Supplementary Appendix 4). Analysis of delta K for A. digitatum suggested K=2; however, the delta K method is known to be unsuitable for accurately identifying K when K=1 (Evanno et al., 2005).

STRUCTURE analysis for Eunicella verrucosa using all populations (a) and hierarchical STRUCTURE analysis using populations from only Britain and France (b). The colours in the STRUCTURE plots correspond to genetic clusters, in which each individual is represented as a coloured vertical bar that represents that individual’s membership in each cluster. See Table 1 for details on population codes.
Analysis of pairwise genetic and geographic distances between sample sites showed a moderate, significant correlation for E. verrucosa (r2=0.348, P=0.001; Figure 4a). The correlation was much weaker but remained significant when the samples from Portugal were excluded from the analysis (r2=0.083, P=0.004; Supplementary Appendix 5). Similarly, the correlation remained significant when the samples from Portugal and Ireland were excluded from the analysis (r2=0.077, P=0.003) (Supplementary Appendix 5). For A. digitatum, a weak, but similarly significant correlation between genetic and geographic distances was apparent (r2=0.045, P=0.035; Figure 4b); however, removal of the North Sea samples resulted in no correlation (r2<0.001, P=0.463; Supplementary Appendix 5). Analysis of both species was also carried out using G”ST as the genetic distance; for E. verrucosa the result was similar to that obtained using FST (Supplementary Appendix 5), however, for A. digitatum, the correlation was lower and non-significant (Supplementary Appendix 5).

Relationship between genetic distance and geographic distance for Eunicella verrucosa (a) and Alcyonium digitatum (b).
Gene flow and effective population size
To estimate gene flow, samples of both species were classified by geographical region as per the AMOVA groupings. Contemporary gene flow estimates (using BayesAss) for both species indicated that the majority of gene flow originated from sample sites within regions (Figure 5). However, for both species, where some gene flow between regions was detected, populations in southwest Britain were the predominant source of exogenous allelic variants. For E. verrucosa, gene flow from Britain was predominantly into France, whereas in A. digitatum gene flow from southwest Britain into the North Sea, Ireland and France was observed. In comparison, contemporary gene flow into Britain appeared very limited for both species. For E. verrucosa, little genetic material was exchanged between Ireland and any other region; likewise, gene flow to/from Portugal was minimal, except for some minor gene flow from Portugal into France. Little or no gene flow from France was detected, suggesting that E. verrucosa in both France and Ireland are effectively sinks. In contrast, for A. digitatum, some gene flow from France to other regions was apparent, although gene flow from the North Sea and Ireland to other study areas was all but absent. For both species, estimates of historical gene flow (using Migrate-n) were somewhat more complex, with populations from Britain again acting as the main source of gene flow for both species, and with only limited gene flow into southwest Britain (Figure 5). Overall, historically, there appeared to have been considerably more gene flow between all regions.

Gene flow diagrams for Eunicella verrucosa and Alcyonium digitatum. Contemporary gene flow estimates were derived from BayesAss and historical gene flow estimates were calculated using Migrate-n. Colours correspond to regions: Britain (red), France (blue), Ireland (green), Portugal (orange), the North Sea (purple). The direction of an arrow represents the direction of gene flow from one region to another. The width of the arrows denotes the relative amount of gene flow within the scenario being explored (that is, the wider the arrow, the more gene flow). The ‘humps’ in the estimates of contemporary gene flow represent gene flow originating from sample sites within regions. Patterns for each diagram are independent, that is, similar widths of arrows or humps do not represent the same amount of gene flow across each of the four diagrams (see Supporting Appendix 6 for exact gene flow estimates).
Analyses of Ne were run using the same groupings as used in the gene flow analyses. Estimates of contemporary effective population sizes were infinite for both species (Supplementary Appendix 6). Historical effective population sizes for E. verrucosa indicated that samples from Britain had the largest Ne, followed by those from Ireland, France and Portugal. In contrast, for A. digitatum, estimates of historical Ne in Ireland and the North Sea were by far the largest, being more than six times larger than the Ne for E. verrucosa in Britain. Estimates of Ne for A. digitatum from Britain and France were, in contrast, very small.
Discussion
This study demonstrates that regional population structure is apparent in the octocoral species E. verrucosa sampled from sites around the northeast Atlantic, including northwest Ireland, southwest Britain, northwest France and southern Portugal. However, over a similar spatial area, another temperate octocoral, A. digitatum, showed only very limited population structure. Therefore, despite the similarities in habitat and life histories of these octocorals, patterns of genetic connectivity over approximately the same geographical area appear variable between species within Octocorallia. The implications of and possible causes for these apparent differences –differences in gene flow and/or effective population size– are now considered.
Genetic diversity and inbreeding
Genetic diversity measures (Hexp and Ar) were generally uniform across the sampling ranges of each species (Tables 1 and 2); however, higher estimates of both measures in A. digitatum indicated higher genetic diversity in this species than in E. verrucosa. In comparison to other studies of temperate corals (Table 3), the genetic diversity of A. digitatum observed in the current study was higher than or comparable to that reported in the octocorals Eunicella singularis (Costantini et al., 2016) and E. cavolini (Masmoudi et al., 2016), and the stony coral Astroides calycularis (Casado-Amezú et al., 2012), but less than two other Mediterranean octocorals, Corallium rubrum (Ledoux et al., 2010) and Paramuricea clavata (Mokhtar-Jamai et al., 2011). In contrast, E. verrucosa exhibited the lowest genetic diversity, a finding that may be explained by both biological/ecological and genetic methodology factors: one highly variable locus, Ever009, which exhibited nine alleles when originally developed (Holland et al., 2013a), was excluded from the current analysis due to the presence of null alleles. At the same time, while the low diversity statistics reflect low polymorphism at some E. verrucosa loci, reduced polymorphism may itself have been the product of an overall lower level of genetic diversity within the populations studied: at four loci (Ever005, Ever008, Ever011 Ever012) only two or three alleles were detected during initial testing (Holland et al., 2013a), with a maximum of five alleles detected at these loci in the current study. The precise biological/ecological causes of this low genetic diversity (for example, inbreeding, selection) remain to be determined. Overall, differences in the patterns of genetic diversity (Hexp and Ar) detected between the two species studied were markedly consistent and may, at least in part, be explained by higher genetic connectivity in A. digitatum.
Eunicella verrucosa has previously been reported as having a low dispersal potential (Munro, 2004); if correct, this would increase the potential for inbreeding. However, the findings of Munro (2004) were based on analysis of only four isoenzymes, markers notorious for their lack of resolution compared to more modern PCR-based techniques (for example, Stevens and Tibayrenc, 1995) and the range and limited number of significant inbreeding coefficients (FIS) observed for E. verrucosa in the current study suggests that the frequency of inbreeding is low, variable between sites and likely due to site-specific factors. Regarding the use of FIS, while the coefficient is typically referred to as measuring the degree of inbreeding within a population, it actually measures homozygosity excess relative to Hardy-Weinberg expectations, and other processes, for example, the inadvertent combining of data from populations with different allele frequencies (the so called ‘Wahlund effect’) can also drive significant positive FIS results. Such a consideration is relevant when seeking to explain the higher number of significant positive FIS values obtained for E. verrucosa populations (Table 1), as this species showed considerably more evidence of genetic structuring (Figure 2a, Supplementary Appendix S2b) than did A. digitatum (Figure 2b, Supplementary Appendix S2d) across the range studied. Thus, given the higher proportion of significant between-population pairwise FSTs (Supplementary Appendix S2b) observed for E. verrucosa, it is possible that cryptic intra-population genetic differentiation may also have played a role in driving significant FIS values in this species. If our FIS results (especially for E. verrucosa) were due to Wahlund effects, such findings would suggest even less inbreeding within the species than the small amount currently postulated. Additionally, the generally low FIS values observed also accord with the low proportion of duplicate genotypes detected in E. verrucosa (<2%) across the study; the number of E. verrucosa individuals at a site with duplicate genotypes ranged from 0 (most samples) to 4 in a small sample (Cam, N=11) from north Cornwall. Interestingly, the north Cornwall sample was one of the few samples not collected by our dive teams, and the relatively high proportion of duplicate genotypes at this site may be a reflection of sampling practice rather than biological reality.
For A. digitatum, significant FIS coefficients were even fewer and lower (though still positive), suggesting only very limited inbreeding in this species; likewise, only a very low proportion (~1%) of all individual A. digitatum successfully genotyped had duplicate profiles. While such findings might be expected for broadcast spawning corals (Ayre and Hughes, 2000), exceptions to this pattern are not uncommon; for example, Combosch and Vollmer (2011) studied populations of Pocillopora damicornis, a broadcast spawning tropical reef coral, and reported a range of large, mostly positive, significant inbreeding coefficients (FIS range: −0.048–0.421), leading them to conclude that widespread inbreeding was apparent in this species in the eastern Pacific.
Compared to previous population genetics studies in octocoral species (for example, Ledoux et al., 2010; Mokhtar-Jamai et al., 2011), the number of significant FIS estimates reported here for E. verrucosa and A. digitatum is globally low: ten significant FIS estimates at 27 sites for E. verrucosa (Table 1) and four significant FIS estimates at 20 sites for A. digitatum (Table 2). Overall, such low estimates of FIS, considered together with the very low numbers of identical individuals sampled in both species, is suggestive of low levels of inbreeding in these two species of octocoral in these parts of their respective ranges.
Genetic structure and connectivity
Population genetic structure was apparent for E. verrucosa at a regional spatial scale (500–2000 km between sample sites), suggesting restrictions to gene flow between populations in different geographical regions. This finding was also supported by analyses of contemporary gene flow (though less so historically), as demonstrated by the limited exchange of genetic material between regions, except for some gene flow between Britain and France (<500 km distance between sites; Figures 3b and 5). Indeed, in both species, analysis of contemporary gene flow suggested that the majority of gene flow occurred between sites within geographical regions, as also observed in Paramuricea clavata, a Mediterranean octocoral (Arizmendi-Mejía et al., 2015). In contrast to E. verrucosa, little regional structure was apparent in A. digitatum, and the only significant differentiation detected was between the samples from the North Sea and those from more westerly areas (>550 km distance between sites); this differentiation appeared to be the product of IBD (as evidenced when comparing the results of IBD analysis with and without North Sea samples of A. digitatum; see Figure 4b and Supplementary Figure S5b) and/or a barrier between the samples of western origin and those from the North Sea. Estimates of contemporary effective population size (Ne) were infinite for both species (Supplementary Appendix 6). Assuming these estimates to be accurate, we found no evidence for disequilibrium caused by genetic drift due to a finite number of parents and, thus, any disequilibrium observed was due to sampling error (Waples and Do, 2008). In contrast, estimates of historical effective population sizes were smaller and variable between regions (Supplementary Appendix 6); this result, together with findings from the corresponding analyses of historical and contemporary gene flow (Figure 5) suggest historical patterns of connectivity were not the same as those observed today.
Overall, our findings suggest that A. digitatum is panmictic across the western part of the sampled range. One possible explanation for this apparent panmixia is that the winter spawning of A. digitatum may facilitate longer dispersal distances via wind-driven currents, thereby increasing genetic connectivity in the eastern Atlantic. Panmixia across similar spatial scales has been reported previously in other marine taxa, including cuttlefish (Wolfram et al., 2006; microsatellite-based study), sea stars (Baus et al., 2005; AFLP-based study), and a closely related species, Alcyonium hibernicum (McFadden, 1999; isoenzyme-based study), although in the latter study, in which little or no genetic variation was detected in A. hibernicum across the Atlantic, McFadden (1999) also linked her findings to high levels of asexual reproduction by parthenogenesis. Similarly, a recent broad study by Gagnaire et al. (2015) highlights the potential impact of large effective population size as an alternative explanation to contemporary panmixia in acting to limit genetic drift, thereby constraining the development of genetic structure, even where gene flow is restricted.
For E. verrucosa, populations from Portugal were differentiated from the majority of populations north of the Bay of Biscay. This may represent a natural break in gene flow in which genetic drift either side of the break is the primary driver of population structure; such a conclusion is supported by both the multivariate (PCoA) and Bayesian clustering (STRUCTURE) analyses. This pattern has been reported previously in a number of taxa, including bivalves (Arias et al., 2010), brittlestars (Muths et al., 2009), crustaceans (Papetti et al., 2005; Remerie et al., 2009), microturbellarians (Casu et al., 2011), macroalgae (Neiva et al., 2014) and fish (Milano et al., 2014). However, the significant correlation between genetic and geographic distances in E. verrucosa in the current study indicates that a proportion of the differentiation observed is likely explained by IBD. Further analysis omitting the Portugal populations suggested that IBD explains some of the genetic differentiation observed between Portugal and all other populations, but much less of the differentiation observed between Britain, Ireland and France. Interestingly, comparisons with other temperate corals (Table 3) suggest that contemporary patterns of population structure appear often to be driven, at least in part, by IBD, which is possibly due to their sedentary life history and their lack of or shorter pelagic larval duration compared to other benthic marine species. In E. verrucosa, analysis of IBD showed no change in significance when the samples from Ireland were removed (Supplementary Appendix 5 and Supplementary Figure S5b), indicating IBD to be a less important driver of population structuring in these Irish samples; such a finding suggests genetic differentiation of these range-peripheral populations is more likely driven by other factors, for example, barriers to gene flow and genetic drift and/or selection. Several previous studies of invertebrates sampled from across this region have also reported genetic differentiation in western Ireland compared to other locations in the northeast Atlantic (Remerie et al., 2009; Casu et al., 2011). These studies attributed this differentiation to recolonisation from relatively northerly refugia that persisted in ice-free coastal areas during the last glacial maximum; however, while Casu et al. explained their findings (reduced genetic diversity in more northerly recolonized populations) by reference to founder effects and low numbers of recolonisers (Hewitt, 1996, 1999), Remerie et al. postulated the higher genetic diversity and heterogeneity they observed in glaciated areas to be suggestive of range persistence during the last glacial maximum. Our findings for E. verrucosa from Ireland (which exhibited the lowest genetic diversity detected in our entire study [Hexp, Ar]) are in line with those of Casu et al. (2011) and, likewise, are suggestive of founder effects following post-glacial recolonisation of suitable northerly habitats by small numbers of recolonisers (Nichols and Hewitt, 1994). A lack of sampling at the southern-most limits of the range of E. verrucosa also makes it difficult to infer the precise origins of the populations in northwest Ireland, as gaps in our knowledge concerning the genetic identity of all possible source populations limits the accuracy of any putative recolonisation hypotheses. Furthermore, to what degree the contemporary distribution of E. verrucosa reflects the extent of the species at the last glacial maximum is unknown, but, to date, its distribution appears not to have extended to areas known to be under ice during the last glacial maximum (Hayward and Ryland, 1995; Hewitt, 1996). In contrast, the distribution of A. digitatum in the northeast Atlantic does not show the same pattern and its present day distribution is considerably more northerly, extending from northern Iberia and the Bay of Biscay up to Iceland and Norway (Hayward and Ryland, 1995). Another possible explanation for the differentiation observed in E. verrucosa from Ireland in the current study is that the effect of selection may be sufficiently strong in northwest Ireland to mitigate the homogenising effect of gene flow. The populations of E. verrucosa found in northwest Ireland are known to be peripheral and inhabit the most northerly limits of the species range (Hayward and Ryland, 1995). Moreover, the lower measures of expected heterozygosity and allelic richness observed in both Irish samples are characteristic of marginal populations, which typically have reduced genetic diversity and can often be under intense selection pressures (Johannesson and André, 2006); our tests for selection identified at least one locus under positive selection. At this stage, however, we do not know which selection pressures, if any, may be acting on these most northerly populations of pink sea fan.
In contrast to the patterns observed between regions, our findings for both octocoral species suggested high gene flow and/or large effective population sizes within regions. For example, for E. verrucosa, little differentiation was observed between the two most distant populations within Britain (Sawtooth and Skomer), implying that the transfer of genetic material can potentially occur up to distances of ~480 km. For A. digitatum, gene flow was evident at an even larger spatial scale, suggesting that genetic material can be transferred greater distances, potentially more than 1050 km (Payne’s Rock—Norfolk). These results suggest that genetic connectivity is high at an intra-regional scale in both species. However, as observed in many marine species with similar life history traits, large effective population size can also act to reduce (or eliminate) genetic structure, sometimes even in situations were gene flow is limited (Gagnaire et al., 2015). Thus, in postulating high gene flow within regions, we need to be mindful of the potential effects of large effective population sizes on genetic structure (or lack of) in these two species.
The hierarchical analysis of E. verrucosa population structure revealed a small degree of genetic differentiation between populations in southwest Britain and northwest France at a distance (~200 km) less than that separating some British populations; however, minimal differentiation was evident for A. digitatum across this area. The effects of mid-channel currents and local near-shore eddies (Dauvin, 2012) on cross-channel larval migration remains to be explored, although previous research has identified a potential genetic break around western Brittany in a number of taxa, including polychaetes (Jolly et al., 2005), nematodes (Wielgoss et al., 2008) and bivalves (Becquet et al., 2012). In this study, the contrast in genetic connectivity across the English Channel may result from differences in the reproductive biology of the two study species. The pelagic larval duration of E. verrucosa is not known, however, evidence from this study suggests this could be shorter than the pelagic larval duration for A. digitatum.
Conservation implications and MPAs
Eunicella verrucosa has been listed under the IUCN red list Vulnerable A1d category since 1996, and is recognised as a species facing a high- to medium-term extinction risk due to exploitation (International Union for Conservation of Nature and Natural Resources (IUCN), 2017). It is also listed as a priority species under the UK Biodiversity Action Plan, the UK response to the prevention of biodiversity loss called for by the 1992 Convention on Biological Diversity; in the Republic of Ireland, France and Portugal it does not currently receive any additional protection beyond its IUCN listing. Several of the MCZs recently designated around southwest Britain (for example, Chesil Beach and Stennis Ledges, The Manacles, and The Isles of Scilly) specifically identify E. verrucosa as a Protected Feature in their designation listing, and 60% of E. verrucosa colonies recorded by diver surveys in southwest Britain fall within areas protected by various other European Union legislation (Pikesley et al., 2016). However, not all of these areas are protected from bottom trawling (for example, The Manacles, Whitsand Bay, Chesil Beach and Stennis Ledges MCZs), suggesting that a large proportion of E. verrucosa in Britain remains vulnerable to anthropogenic disturbance and the current level of protection of UK marine ecosystems afforded by the MCZ network is generally insufficient (for example, Lieberknecht and Jones, 2016; Pikesley et al., 2016). Moreover, while the UK government appears to have moved away from the recommended ecological network guidelines for MCZ designation (Lieberknecht and Jones, 2016), the E. verrucosa data presented here highlight interesting findings relative to the conservation of ecologically important and prevalent sessile taxa at local (that is, single-site MPAs) to regional (that is, connected metapopulation) scales.
More specifically, the genetic distinctiveness of E. verrucosa populations from Ireland underpins an argument for protecting particular sites. Marginal populations often contain rare alleles (the highest extent of private alleles were found at these sites), but may recruit more slowly, and may be genetically isolated, implying vulnerability and reduced resilience (Sanderson, 1996) and therefore an increased need for protection. However, away from the edges of the species range, our data suggest that connectivity can be maintained between populations of these species in some designated MCZs. Moreover, the range of E. verrucosa in Britain is small compared to its (primarily Lusitanean) global distribution and, although contemporary connectivity between British populations appears to be a high, at regional spatial scales it could be argued that the genetic distinction of these populations, coupled with their possible role as source populations that act to maintain broader connectivity across this area of the northeast Atlantic, may be sufficient to warrant international conservation efforts (for example, OSPAR Convention, 2013).
In the UK, A. digitatum has no specific protective status, is not at the periphery of its global range, and, in our study (apart from the North Sea—English Channel/eastern Atlantic divide), it exhibited relatively high genetic diversity with little evidence of any major barriers to gene flow. Overall, coupled with the high prevalence of this species in UK waters, these factors imply that this species may be a low priority for protection in its own right, and it is likely to receive only patchy, incidental protection based upon the location of current MCZs designated on the basis of other features, although arguments for consideration of this species in design guidelines could still fit both the ‘representativity’ and ‘replication’ principles (Natural England, 2010). However, reduced heterozygosity and impaired sexual reproduction have been reported in another cnidarian species subjected to trawling damage (Henry and Kenchington, 2004) and reduced colony numbers and size have been reported for A. digitatum in Lyme Bay, southern England, in trawled areas (Hinz et al., 2011); therefore, this species may be locally vulnerable. Certainly, the occurrence of damaged, sessile populations in disturbed areas, are a useful proxy to highlight degraded ecosystems that may also contain more directly threatened species.
The results from the present study suggest that populations of E. verrucosa would benefit from protection across the species range as a connected metapopulation. Although implementing protective measures for a single species across its entire UK range is highly unlikely given commercial and economic pressures within the region, our study serves to highlight areas for consideration in an ecosystem-based management approach. Irish populations of E. verrucosa may warrant protection because of their marginality, yet they are not currently protected within the Republic of Ireland beyond their IUCN listing. Analysis of gene flow for both octocoral species studied suggests populations in southwest Britain act as a source for surrounding regions, highlighting the value in protecting these populations. In the UK, the current recommendation for the spacing of designated MPAs is in the region of 40–80 km (Natural England, 2010). In light of our findings, it appears that the distances between these MPAs would generally be sufficient to maintain genetic connectivity of these two octocoral species in UK waters. Of course, this assumes that contemporary local oceanic currents are able to facilitate the transport of enough larvae in each species, whether by a continuing stepping-stone process or a single dispersal event. Managing effective conservation of marine species with overlapping generations and high levels of clonality, such as sponges and corals, can be challenging because characteristic genotypes may persist for decades to centuries, even after significant barriers to gene flow arise. As a result, traditional F-statistics may not always represent current patterns of genetic connectivity (Botsford et al., 2009) and these factors should be incorporated when including genetic data into MPA network designation. Furthermore, because of the challenges associated with genotyping octocorals, such as the slow rate of mitochondrial evolution (McFadden et al., 2010) and the difficulty of isolating microsatellites (Liu et al., 2005), the type and numbers of molecular marker used may not be powerful enough to detect a signal of fine-scale population structure. As seen in the current study, while relatively strong patterns of regional structure were detected in E. verrucosa, except for some weak structuring between French and English samples of E. verrucosa in the Channel, no fine-scale structure (<200 km between sample sites) was detected for either of the species studied. This may be indicative of genetic connectivity between these populations, but could also represent a lack of power in the genetic markers used. Exploration of alternative marker systems may deliver improved resolution (for example, Shinzato et al., 2015) and should prove valuable for future conservation research and the management of MPA networks.
In conclusion, genetic diversity appears to be uniform across the range studied in both species; however, genetic diversity was low in E. verrucosa, whereas in A. digitatum, it was slightly higher, but still lower than that reported for two species of Mediterranean octocorals (Ledoux et al., 2010; Mokhtar-Jamai et al., 2011). For both species, only limited inbreeding was apparent, and whether this has an impact on fitness and long-term resilience of the populations in question is currently unknown. Regional population structure was identified in E. verrucosa, indicative of departures from panmixia at large spatial scales; in contrast, in A. digitatum, apart from some genetic differentiation between populations from the North Sea and those from the English Channel/eastern Atlantic, we found little population structure, suggesting high gene flow and connectivity in this species in the western part of the range sampled. Contemporary and historical estimates of effective population size were contrasting and generally difficult to interpret, and for both species the potential role of large Nes in masking a lack of gene flow cannot be ruled out. Patterns of gene flow were complex, but indicated Britain as a source of genetic variants for both species. Several populations of both species are represented in the UK MPA network and, given the ecological importance of both species, continued monitoring and assessment of their genetic diversity within and beyond protected sites could be a useful measure of the efficacy of the existing network, and a valuable guide to the designation of new MCZs.
Data archiving
Microsatellite genotypes for Eunicella verrucosa and Alcyonium digitatum are available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.nj0v4.
References
Almany GR, Connolly SR, Heath DD, Hogan JD, Jones GP, McCook LJ et al. (2009). Connectivity, biodiversity conservation and the design of marine reserve networks for coral reefs. Coral Reefs 28: 339–351.
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G . (2008). LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics 9: 323.
Arias A, Fernández-Moreno M, Fernández-Tajes J, Gaspar MB, Méndez J . (2010). Strong genetic differentiation among east Atlantic populations of the sword razor shell (Ensis siliqua assessed with mtDNA and RAPD markers. Helgoland Mar Res 65: 81–89.
Arizmendi-Mejía R, Linares C, Garrabou J, Antunes A, Ballesteros E, Cebrian E, Díaz D, Ledoux J-B . (2015). Combining genetic and demographic data for the conservation of a mediterranean marine habitat-forming species. PLoS One 10: e0119585.
Aurelle D, Ledoux J-B, Rocher C, Borsa P, Chenuil A, Féral J-P . (2011). Phylogeography of the red coral (Corallium rubrum: inferences on the evolutionary history of a temperate gorgonian. Genetica 139: 855–869.
Ayre DJ, Hughes TP . (2000). Genotypic diversity and gene flow in brooding and spawning corals along the Great Barrier Reef, Australia. Evolution 54: 1590–1605.
Bargelloni L, Alarcon JA, Alvarez MC, Penzo E, Magoulas A, Palma J et al. (2005). The Atlantic-Mediterranean transition: discordant genetic patterns in two seabream species, Diplodus puntazzo and Diplodus sargus. Mol Phylogenet Evol 36: 523–535.
Bargelloni L, Alarcon JA, Alvarez MC, Penzo E, Magoulas A, Reis C et al. (2003). Discord in the family Sparidae Teleostei: divergent phylogeographical patterns across the Atlantic-Mediterranean divide. J Evol Biol 16: 1149–1158.
Baus E, Darrock DJ, Bruford MW . (2005). Gene-flow patterns in Atlantic and Mediterranean populations of the Lusitanian sea star Asterina gibbosa. Mol Ecol 14: 3373–3382.
Becquet V, Simon-Bouhet B, Pante E, Hummel H, Garcia P . (2012). Glacial refugium versus range limit: conservation genetics of Macoma balthica, a key species in the Bay of Biscay (France). J Exp Mar Bio Ecol 432–433: 73–82.
Beerli P, Felsenstein J . (2001). Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci USA 98: 4563–4568.
Botsford LW, White JW, Coffroth MA, Paris CB, Planes S, Shearer TL et al. (2009). Connectivity and resilience of coral reef metapopulations in marine protected areas: matching empirical efforts to predictive needs. Coral Reefs 28: 327–337.
Casado-Amezú P, Goffredo S, Templado J, MacHordom A . (2012). Genetic assessment of population structure and connectivity in the threatened mediterranean coral Astroides calycularis Scleractinia, Dendrophylliidae at different spatial scales. Mol Ecol 21: 3671–3685.
Casu M, Sanna D, Cossu P, Lai T, Francalacci P, Curini-Galletti M . (2011). Molecular phylogeography of the microturbellarian Monocelis lineata (Platyhelminthes: Proseriata) in the North-East Atlantic. Biol J Linn Soc 103: 117–135.
Charrier G, Chenel T, Durand JD, Girard M, Quiniou L, Laroche J . (2006). Discrepancies in phylogeographical patterns of two European anglerfishes (Lophius budegassa and Lophius piscatorius. Mol Phylogenet Evol 38: 742–754.
Combosch DJ, Vollmer SV . (2011). Population genetics of an ecosystem-defining reef coral Pocillopora damicornis in the Tropical Eastern Pacific. PLoS One 6: e21200.
Costantini F, Fauvelot C, Abbiati M . (2007). Genetic structuring of the temperate gorgonian coral (Corallium rubrum across the western Mediterranean Sea revealed by microsatellites and nuclear sequences. Mol Ecol 16: 5168–5182.
Costantini F, Gori A, Lopez-González P, Bramanti L, Rossi S, Gili J-M et al. (2016). Limited genetic connectivity between gorgonian morphotypes along a depth gradient. PLoS One 11: e0160678.
Cowen RK, Gawarkiewicz G, Pineda J, Thorrold SR, Werner FE . (2007). Population connectivity in marine systems. Oceanography 20: 14–21.
Cowen RK, Sponaugle S . (2009). Larval dispersal and marine population connectivity. Ann Rev Mar Sci 1: 443–466.
Da Silva AG, Appleyard SA, Upston J . (2014). Establishing the evolutionary compatibility of potential sources of colonizers for overfished stocks: a population genomics approach. Mol Ecol 24: 564–579.
Dauvin JC . (2012). Are the eastern and western basins of the English Channel two separate ecosystems? Mar Poll Bull 64: 463–471.
Dunn OJ . (1961). Multiple comparisons among means. J Am Stat Assoc 56: 52–64.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Lischer HEL . (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10: 564–567.
Fogarty MJ, Botsford LW . (2007). Population connectivity and spatial management of marine fisheries. Oceanography 20: 112–123.
Foster NL, Paris CB, Kool JT, Baums IB, Stevens JR, Sanchez JA et al. (2012). Connectivity of Caribbean coral populations: complementary insights from empirical and modelled gene flow. Mol Ecol 21: 1143–1157.
Francis RM . (2017). POPHELPER: an R package and web app to analyse and visualise population structure. Mol Ecol Resour 17: 27–32.
Gagnaire P-A, Broquet T, Aurelle D, Viard F, Souissi A, Bonhomme F, Arnaud-Haond S, Bierne N . (2015). Assessing connectivity of marine populations in the genomic era. Evol Appl 8: 769–786.
Gerber LR, Del Mark Mancha-Cisneros M, O’Connor M, Selig ER . (2014). Climate change impacts on connectivity in the ocean: implications for conservation. Ecosphere 5: 1–18.
Goffredo S, Di Ceglie S, Zaccanti F . (2009). Genetic differentiation of the temperate-subtropical stony coral Leptopsammia pruvoti in the Mediterranean Sea. Isr J Ecol Evol 55: 99–115.
Hall-Spencer JM, Pike J, Munn CB . (2007). Diseases affect cold-water corals too: Eunicella verrucosa (Cnidaria: Gorgonacea) necrosis in SW England. Diseases of Aquatic Organisms 76: 87–97.
Hartnoll RG . (1975). The annual cycle of Alcyonium digitatum. Estuar Coast Mar Sci 3: 71–78.
Hayward PJ, Ryland JS . (1995) Handbook of the Marine Fauna of North-west Europe. Oxford University Press: Oxford.
Hedgecock D, Barber PH, Edmands S . (2007). Genetic approaches to measuring connectivity. Oceanography 20: 70–79.
Henry LA, Kenchington ELR . (2004). Ecological and genetic evidence for impaired sexual reproduction and induced clonality in the hydroid Sertularia cupressina Cnidaria Hydrozoa on commercial scallop grounds in Atlantic Canada. Marine Biology 145: 1107–1118.
Herrera S, Shank TM, Sánchez JA . (2012). Spatial and temporal patterns of genetic variation in the widespread antitropical deep-sea coral Paragorgia arborea. Mol Ecol 21: 6053–6067.
Hewitt GM . (1996). Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58: 247–276.
Hewitt GM . (1999). Post-glacial re-colonization of European biota. Biol J Linn Soc 68: 87–112.
Hinz H, Tarrant D, Ridgeway A, Kaiser MJ, Hiddink JG . (2011). Effects of scallop dredging on temperate reef fauna. Mar Ecol Prog Ser 432: 91–102.
Hiscock K, Sharrock S, Highfield J, Snelling D . (2010). Colonization of an artificial reef in south-west England-ex-HMS ‘Scylla’. J Mar Biol Assoc UK 90: 69–94.
Holland LP, Dawson DA, Horsburgh GJ, Krupa AP, Stevens JR . (2013a). Isolation and characterization of fourteen microsatellite loci from the endangered octocoral Eunicella verrucosa (Pallas, 1766). Conserv Genet Resour 5: 825–829.
Holland LP, Dawson DA, Horsburgh GJ, Stevens JR . (2013b). Isolation and characterization of 11 microsatellite loci from the ubiquitous temperate octocoral Alcyonium digitatum (Linnaeus, 1758). Conserv Genet Resour 5: 767–770.
International Union for Conservation of Nature and Natural Resources (IUCN). (2017). Eunicella verrucosa (Broad Sea Fan, Pink Sea Fan). Available at: http://dx.doi.org/10.2305/IUCN.UK.1996.RLTS.T8262A12903486.en (accessed on 15 December 2016).
Johannesson K, André C . (2006). Life on the margin: genetic isolation and diversity loss in a peripheral marine ecosystem, the Baltic Sea. Mol Ecol 15: 2013–2029.
Jolly MT, Jollivet D, Gentil F, Thiébaut E, Viard F . (2005). Sharp genetic break between Atlantic and English Channel populations of the polychaete Pectinaria koreni, along the north coast of France. Heredity 94: 23–32.
Jones GP, Srinivasan M, Almany GR . (2007). Population connectivity and conservation of marine biodiversity. Oceanography 20: 100–111.
Kalinowski ST . (2005). HP-RARE 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5: 187–189.
Kalinowski ST, Taper ML, Marshall TC . (2007). Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16: 1099–1106.
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA . (2013). diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4: 782–788.
Kool JT, Moilanen A, Treml EA . (2013). Population connectivity: recent advances and new perspectives. Landsc Ecol 28: 165–185.
Ledoux JB, Mokhtar-Jamai K, Roby C, Feral JP, Garrabou J, Aurelle D . (2010). Genetic survey of shallow populations of the Mediterranean red coral [Corallium rubrum (Linnaeus, 1758)]: new insights into evolutionary processes shaping nuclear diversity and implications for conservation. Mol Ecol 19: 675–690.
Lieberknecht LM, Jones PJS . (2016). From stormy seas to the doldrums: the challenges of navigating towards an ecologically coherent marine protected area network through England’s Marine Conservation Zone process. Marine Policy 71: 275–284.
Liu SYV, Dai CF, Fan TY, Yu HT . (2005). Cloning and characterization of microsatellite loci in a gorgonian coral, Junceella juncea (Anthozoa; Octocorallia Ellisellidae and its application in clonal genotyping. Mar Biotechnol 7: 26–32.
Lowe WH, Allendorf FW . (2010). What can genetics tell us about population connectivity? Molecular Ecology 19: 3038–3051.
Marti-Puig P, Costantini F, Rugiu L, Ponti M, Abbiati M . (2013). Patterns of genetic connectivity in invertebrates of temperate MPA networks. Adv Oceanogr Limnol 4: 138–149.
Masmoudi MB, Chaoui L, Topçu NE, Hammami P, Kara MH, Aurelle D . (2016). Contrasted levels of genetic diversity in a benthic Mediterranean octocoral: consequences of different demographic histories? Ecol Evol 6: 8665–8678.
McCook LJ, Almany GR, Berumen ML, Day JC, Green AL, Jones GP et al. (2009). Management under uncertainty: guide-lines for incorporating connectivity into the protection of coral reefs. Coral Reefs 28: 353–366.
McFadden C . (1999). Genetic and taxonomic relationships among northeastern Atlantic and Mediterranean populations of the soft coral Alcyonium coralloides. Mar Biol 133: 171–184.
McFadden CS, Donahue R, Hadland BK, Weston R . (2001). A molecular phylogenetic analysis of reproductive trait evolution in the soft coral genus. Alcyonium. Evolution 55: 54–67.
McFadden CS, Hutchinson MB . (2004). Molecular evidence for the hybrid origin of species in the soft coral genus Alcyonium (Cnidaria: Anthozoa: Octocorallia). Molecular Ecology 13: 1495–1505.
McFadden CS, Sánchez JA, France SC . (2010). Molecular phylogenetic insights into the evolution of Octocorallia: a review. Integr Comp Biol 50: 389–410.
Meirmans PG, Hedrick PW . (2011). Assessing population structure: FST and related measures. Mol Ecol Resour 11: 5–18.
Milano I, Babbucci M, Cariani A, Atanassova M, Bekkevold D, Carvalho GR et al. (2014). Outlier SNP markers reveal fine-scale genetic structuring across European hake populations (Merluccius merluccius. Mol Ecol 23: 118–135.
Mokhtar-Jamai K, Pascual M, Ledoux JB, Coma R, Feral JP, Garrabou J et al. (2011). From global to local genetic structuring in the red gorgonian Paramuricea clavata: the interplay between oceanographic conditions and limited larval dispersal. Mol Ecol 20: 3291–3305.
Mora C, Sale P . (2011). Ongoing global biodiversity loss and the need to move beyond protected areas: a review of the technical and practical shortcomings of protected areas on land and sea. Mar Ecol Prog Ser 434: 251–266.
Munday P, Leis J, Lough J, Paris C, Kingsford M, Berumen M, Lambrechts J . (2009). Climate change and coral reef connectivity. Coral Reefs 28: 379–395.
Munro L . (2004). Determining the reproductive cycle of Eunicella verrucosa. Reef Res. RR Report 07/2004 ETR 12, p28.
Muths D, Jollivet D, Gentil F, Davoult D . (2009). Large-scale genetic patchiness among NE Atlantic populations of the brittle star Ophiothrix fragilis. Aquat Biol 5: 117–132.
Natural England (2010). Guidance on the size and spacing of Marine Protected Areas in England. Natural England Report (NECR037), ISSN 2040-5545. Natural England, Northminster House: Peterborough, UK.
Neiva J, Assis J, Fernandes F, Pearson GA, Serrão EA . (2014). Species distribution models and mitochondrial DNA phylogeography suggest an extensive biogeographical shift in the high-intertidal seaweed Pelvetia canaliculata. J Biogeogr 41: 1137–1148.
Nichols RA, Hewitt GM . (1994). The genetic consequences of long distance dispersal during colonization. Heredity 72: 312–317.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P . (2004). MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
OSPAR Commission. (2006). Overview of OSPAR assessments 1998–2006. OSPAR Commission, London, UK, Publication No.: 287/2006, ISBN 1-905859-25-2 / 978-1-905859-25-2.
OSPAR Convention. (2013). An assessment of the Ecological coherence of the OSPAR Network of Marine Protected Areas in 2012. OSPAR Commission, London, UK, ISBN 978-1-909159-52-5.
Papetti C, Zane L, Bortolotto E, Bucklin A, Patarnello T . (2005). Genetic differentiation and local temporal stability of population structure in the euphausiid Meganyctiphanes norvegica. Mar Ecol Prog Ser 289: 225–235.
Palumbi SR . (2003). Population genetics, demographic connectivity, and the design of marine reserves. Ecol Appl 13: S146–S158.
Peakall R, Smouse P . (2012). GenAlEx 6.5 : Genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 1: 6–8.
Pérez-Portela R, Turon X, Bishop J . (2012). Bottlenecks and loss of genetic diversity: spatio-temporal patterns of genetic structure in an ascidian recently introduced in Europe. Mar Ecol Prog Ser 451: 93–105.
Pikesley SK, Godley BJ, Latham H, Richardson PB, Robson LM, Solandt J-L et al. (2016). Pink sea fans (Eunicella verrucosa as indicators of the spatial efficacy of marine protected areas in Southwest UK coastal waters. Mar Policy 64: 38–45.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Puckett BJ, Eggleston DB, Kerr PC, Luettich RA . (2014). Larval dispersal and population connectivity among a network of marine reserves. Fish Oceanogr 23: 342–361.
Rambaut A, Suchard M, Xie D, Drummond A . (2014). Tracer v1.6. Available at: http://beast.bio.ed.ac.uk/Tracer.
R Development Core Team. (2016). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0. Available at: https://www.R-project.org/.
Remerie T, Vierstraete A, Weekers PHH, Vanfleteren JR, Vanreusel A . (2009). Phylogeography of an estuarine mysid, Neomysis integer (Crustacea, Mysida, along the north-east Atlantic coasts. J Biogeogr 36: 39–54.
Rousset F . (2008). GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8: 103–106.
Sanderson WG . (1996). Rarity of marine benthic species in Great Britain: Development and application of assessment criteria. Aquat Conserv 6: 245–256.
Shinzato C, Mungpakdee S, Arakaki N, Satoh N . (2015). Genome-wide SNP analysis explains coral diversity and recovery in the Ryukyu Archipelago. Sci Rep 5: 18211.
Storey JD, Tibshirani R . (2003). Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100: 9440–9445.
Stevens JR, Tibayrenc M . (1995). Detection of linkage disequilibrium in Trypanosoma brucei isolated from tsetse flies and characterized by RAPD analysis and isoenzymes. Parasitology 110: 181–186.
Waples RS, Do C . (2008). LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8: 753–756.
Watling L, Auster PJ . (2005) Distribution of deep-water Alcyonacea off the northeast coast of the United States. In: Freiwald A, Roberts JM (eds). Cold-Water Corals and Ecosystems. Springer-Verlag: Berlin, Heidelberg, Germany, pp 279–296.
Weir BS, Cockerham CC . (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Wielgoss S, Taraschewski H, Meyer A, Wirth T . (2008). Population structure of the parasitic nematode Anguillicola crassus, an invader of declining North Atlantic eel stocks. Mol Ecol 17: 3478–3495.
Wilson GA, Rannala B . (2003). Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163: 1177–1191.
Wolfram K, Mark FC, John U, Lucassen M, Pörtner HO . (2006). Microsatellite DNA variation indicates low levels of genetic differentiation among cuttlefish (Sepia officinalis populations in the English Channel and the Bay of Biscay. Comp Biochem Physiol D 1: 375–383.
Acknowledgements
We thank D Laffoley (Natural England, IUCN) and J Ashworth (Natural England) for invaluable advice and help with project conception and planning. We are grateful to the following for assistance with sample collection: J Kent and S Williams (University of Bristol Underwater Club), P Shears and J Shears (University of Exeter), J Turner (University of Exeter Sub-Aqua Club), J Hall-Spencer and M Whelan (Plymouth University), K Hiscock (Marine Biological Association UK), P Newman (Countryside Council for Wales), C Munro (Marine Bio-images, Exeter), R Jones (Zoological Society of London), I Reach (Natural England), B Pearce and J Pinnion (Marine Ecological Surveys), P Romans and staff (Observatoire Océanologique de Banyuls, France), L Lévêque and staff (Station Biologique de Roscoff, France) and R Castilho and staff (University of Algarve, Portugal). Sampling within the UK was carried out under Natural England licences 20080861 and 20090943, and Marine Management Organisation licence MMO-0001. Genotyping was performed at the Natural Environment Research Council, UK (NERC) Biomolecular Analysis Facility, University of Sheffield and we thank D Dawson, G Horsburgh and A Krupa for technical assistance. We thank Natural England (project No. RP0286, contract No. SAE 03-02-146), the NERC (grant No. NE/L002434/1) and the University of Exeter for funding this research. Additional funding for sample collection, travel and microsatellite development was provided by the EU Framework 7 ASSEMBLE programme, agreement no. 227799, and NERC grant No. NBAF-362. We thank the editor and three reviewers for their helpful comments in revising this manuscript. This research forms part of the PhDs of LPH and TLJ. LPH is currently at the School of Biological Sciences, Victoria University of Wellington, New Zealand.
Author contributions
LPH conducted laboratory analysis, carried out sample collection, undertook preliminary analyses and wrote the manuscript. TLJ analysed the data and wrote the manuscript. JRS conceived, designed and directed the study, carried out sample collection and wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Holland, L., Jenkins, T. & Stevens, J. Contrasting patterns of population structure and gene flow facilitate exploration of connectivity in two widely distributed temperate octocorals. Heredity 119, 35–48 (2017). https://doi.org/10.1038/hdy.2017.14
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2017.14
This article is cited by
-
Novel species discrimination method elucidates colony growth form variation between the New Zealand soft corals Kotatea aurantiaca and Kotatea lobata
Marine Biodiversity (2023)
-
Population genetic structure of the deep-sea precious red coral Hemicorallium laauense along the Hawaiian Ridge
Marine Biology (2023)
-
Genetic homogeneity, lack of larvae recruitment, and clonality in absence of females across western Mediterranean populations of the starfish Coscinasterias tenuispina
Scientific Reports (2021)
-
Outlier SNPs detect weak regional structure against a background of genetic homogeneity in the Eastern Rock Lobster, Sagmariasus verreauxi
Marine Biology (2018)
