
Abstract
Allopolyploidization often happens recurrently, but the evolutionary significance of its iterative nature is not yet fully understood. Of particular interest are the gene flow dynamics and the mechanisms that allow young sibling polyploids to remain distinct while sharing the same ploidy, heritage and overlapping distribution areas. By using eight highly variable nuclear microsatellites, newly reported here, we investigate the patterns of divergence and gene flow between 386 polyploid and 42 diploid individuals, representing the sibling allopolyploids Dactylorhiza majalis s.s. and D. traunsteineri s.l. and their parents at localities across Europe. We make use in our inference of the distinct distribution ranges of the polyploids, including areas in which they are sympatric (that is, the Alps) or allopatric (for example, Pyrenees with D. majalis only and Britain with D. traunsteineri only). Our results show a phylogeographic signal, but no clear genetic differentiation between the allopolyploids, despite the visible phenotypic divergence between them. The results indicate that gene flow between sibling Dactylorhiza allopolyploids is frequent in sympatry, with potential implications for the genetic patterns across their entire distribution range. Limited interploidal introgression is also evidenced, in particular between D. incarnata and D. traunsteineri. Altogether the allopolyploid genomes appear to be porous for introgression from related diploids and polyploids. We conclude that the observed phenotypic divergence between D. majalis and D. traunsteineri is maintained by strong divergent selection on specific genomic areas with strong penetrance, but which are short enough to remain undetected by genotyping dispersed neutral markers.
Similar content being viewed by others
Introduction
Allopolyploidization is the main conduit of reticulate evolution in angiosperms, bringing together two or more diverged genomes into a single nucleus (Comai et al., 2003; Otto, 2007; Soltis and Soltis, 2009). The novel genomic status instantly triggers genetic and epigenetic responses, mostly of a stochastic nature (Paun et al., 2007; Madlung and Wendel, 2013). These genomic alterations can have dramatic phenotypic implications, including reshaping the ecological attributes of the polyploid individuals (Soltis et al., 2010). Upon formation, neopolyploids are strongly filtered by natural selection towards stabilizing meiosis and towards increased fitness in their respective habitats. This is an important point, as allopolyploids often form repeatedly between different populations of the same parental species, potentially resulting in an array of substantially different lineages (Soltis and Soltis, 2009) due to (i) origins from different parental genotypes, (ii) chance genomic alterations after allopolyploidization, (iii) different selection pressures that the neopolyploids may experience and/or (iv) difference in extent/direction of backcrossing towards diploid parental species. However, the long-term evolutionary significance of recurrent allopolyploidization remains to date not completely understood (Doyle et al., 2004; Meimberg et al., 2009; Soltis et al., 2010).
Multiple origins intuitively increase the probability that the polyploid cytotype will succeed to become established. In addition, they create the opportunity for increased genetic and ecological breadth of the polyploid cytotype. Under isolation, sibling allopolyploids could in time evolve as independent species (Perrie et al., 2010; Chester et al., 2012; Levin, 2013). However, the distribution of sibling allopolyploid lineages may partly overlap and gene flow is likely to occur (‘lineage recombination’ sensu, Doyle et al., 1999). Gene flow between sibling allopolyploids may augment their genetic variability and further enhance the evolutionary success of sexual allopolyploid species (Meimberg et al., 2009; Modliszewski and Willis, 2012). On the other hand, lineage recombination could result in homogenization of sibling allopolyploids, counteracting diversification.
For the sibling allopolyploids to survive as independent units, it appears therefore important that some degree of reproductive isolation among them develops early. Mating barriers could ensue as chance alternative silencing or physical loss of homeologs, resulting in hybrid lethality or sterility (‘Oka model’; Oka, 1953; Lynch and Force, 2000; Bikard et al., 2009). Furthermore, mechanisms such as ‘divergence hitchhiking’ and ‘genome hitchhiking’ could act and allow reproductive isolation to develop in time despite genome homogenization by gene flow (Via, 2012; Feder et al., 2012). From an evolutionary point of view, such sibling allopolyploids could be compared with sister taxa evolving by speciation-with-gene-flow (Nosil et al., 2009). Clearly, a proper understanding of gene flow dynamics between young sibling polyploids is a key to reveal the evolutionary significance of the iterative nature of polyploidization.
An excellent system to study the early evolution of natural sibling allopolyploids is provided by the Dactylorhiza majalis complex (Orchidaceae). Dactylorhiza constitutes an unusually dynamic system of hybridization between diploids, coupled with polyploid formation. Allotetraploid taxa (2n=80) of the D. majalis complex (Hedrén, 1996) have arisen by multiple, independent origins from unidirectional hybridization between the same diploid parental lineages (D. fuchsii, always acting as the maternal parent, and D. incarnata; both 2n=40; Pillon et al., 2007). Hybridization and introgression between independently derived allotetraploids has been frequently reported (for example, Hedrén, 1996, 2003; Hedrén et al., 2001, 2011). It is therefore of interest to understand the mechanisms that allow these young sibling allopolyploids to remain distinct over time, even in sympatry, while sharing the same genetic heritage and ploidy (Paun et al., 2011).
Many studies of allopolyploid evolution have previously considered either old polyploid complexes in which parental lineages have been lost and the allopolyploid derivatives have acquired distinct molecular profiles by extensive post-polyploidization evolution (for example, Shaked et al., 2001; Chaudhary et al., 2009; Marcussen et al., 2010), or else considered young complexes in which no/little intermixing of independently derived neopolyploids have yet occurred (for example, Soltis et al., 2004). In the present study, we investigate the genetic structure and the patterns of gene flow between two medium-aged Dactylorhiza allopolyploids at different localities across Europe by using highly-polymorphic simple sequence repeats markers (SSR or microsatellites) as a basis for discussing the possible outcomes of admixture between sibling allopolyploids. Our study addresses the following specific questions: (1) Are the sibling Dactylorhiza allopolyploids genetically differentiated? (2) Are there signals of gene flow between the sibling allopolyploids and if so, at what scale of geographic distances does this happen? and (3) Are there signals of backcrossing among polyploids and their diploid parental species?
Materials and methods
Study system
We focus here on two widespread allotetraploids of the complex, Dactylorhiza majalis s.s. (Rchb.) P.F. Hunt & Summerh. and D. traunsteineri s.l. (Saut. ex Rchb.) Soó, the latter including D. ebudensis (Wiefelspütz ex R.M. Bateman & Denholm) P. Delforge, following Bateman and Denholm (2012), and D. lapponica (Laest.) Soó. In their characteristic forms, the species have distinct morphology, ecology and geographic distribution (Paun et al., 2010). They can occur in close proximity (in particular in the Alps), but in different microhabitats still separated by at least 100 m. Dactylorhiza majalis s.s. has a wider ecological tolerance and grows in moist meadows on calcareous ground kept open by grazing or mowing. It is characterized by robust and broad-leaved plants with a dense, many-flowered inflorescence (Baumann and Künkele, 1988; Delforge, 2001). Its distribution reaches from the Pyrenees, northern Italy and northern Balkans in the south to Denmark and southernmost Sweden in the north and from France in the west to Poland and the Baltic states in the east (Hultén and Fries, 1986, and revised according to unpublished data). Dactylorhiza traunsteineri is less competitive and grows in calcareous fens, typically on naturally open sites associated with seepage zones, but it also benefits from grazing or mowing (Kull and Hutchings, 2006). It is slender and usually low-grown with narrow leaves and a lax few-flowered inflorescence (Soó, 1980; Baumann and Künkele, 1988; Delforge, 2001). It is relatively widespread in north-western and northern Europe outside the range of D. majalis, but it has more of a localized and disjunct distribution in Central Europe (Hultén and Fries, 1986).
Molecular studies suggest that the two allotetraploids may have been independently formed at different times during the last part of the Quaternary (for example, Pillon et al., 2007). Dactylorhiza majalis appears to be older and to have survived in one or few neighbouring refugia in Central Europe during the last ice age. Dactylorhiza traunsteineri is more heterogeneous and may include younger elements than D. majalis. The wide and scattered distribution of D. traunsteineri and the regional mosaic of plastid DNA variants in the species suggest that it is formed shortly after the last glaciation, or that it survived the last ice age in several periglacial refugia, some also at relatively high latitudes (Nordström and Hedrén, 2009).
The two species contain a number of distinct plastid DNA variants not found in present-day representatives of the maternal D. fuchsii lineage (Pillon et al., 2007; Nordström and Hedrén, 2008, 2009). Accordingly, some degree of shared evolutionary history of the two allopolyploids is suggested as several of these polyploid-specific haplotypes are widespread and common in both tetraploid species. In addition, no clear-cut genetic differentiation among the allopolyploids of the Dactylorhiza majalis complex has been uncovered by investigating ITS sequences (Pillon et al., 2007) and amplified fragment length polymorphism (Hedrén et al., 2001). However, recent studies using cDNA-amplified fragment length polymorphisms (Paun et al., 2011) and methylation-sensitive amplified fragment length polymorphisms (Paun et al., 2010) indicated that Dactylorhiza majalis and D. traunsteineri show some distinct epigenetic and gene expression patterns, most probably as a result of divergent selection acting on the variation released with the allopolyploidization events (Paun et al., 2010, 2011), but it is still an open question whether any genomic differentiation matching these divergent patterns in gene expression is to be found. As Dactylorhiza allopolyploids are at a relatively early stage of evolution (Devos et al., 2006; Pillon et al., 2007; Nordström and Hedrén, 2009) but feature broad and partly overlapping distribution ranges, we hypothesize that extensive lineage recombination involving occasional interbreeding between the two sibling allopolyploids has taken place in the recent past, on the background of a limited genetic divergence between them.
Dactylorhiza diploids and polyploids are mostly bumblebee-pollinated by food deception (Sletvold et al., 2010), rendering pollinator-mediated gene flow between patches higher than in rewarding orchids (Cozzolino and Widmer, 2005). Still, several studies directly monitoring orchid pollen dispersal could only rarely show pollen transport distances larger than 50 m (Kropf and Renner, 2008 and references therein). The dust-like seeds are wind dispersed (Arditti and Ghani, 2000), and, as in other orchids (Squirrel et al., 2001; Cozzolino et al., 2003), a proportion of the overall gene flow is likely to be due to seed dispersal.
Plant sampling
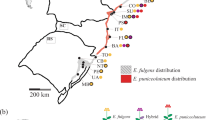
During the summers of 2011–2013, we sampled 386 polyploid individual plants from the Dactylorhiza majalis complex, including the sibling allotetraploids D. traunsteineri s.l. and D. majalis s.s. We studied a total of 24 sampling localities (hereafter referred to as populations) from across the distribution range of this complex in Europe, focusing on Pyrenees, Alps, Scandinavia and Britain (Figure 1, Table 1). The sampling areas of D. majalis and D. traunsteineri were classified into three categories with regard to the proximity of one species to the other: (i) allopatric (Pyrenean and Scandinavian populations of D. majalis versus the British and Scandinavian populations of D. traunsteineri), (ii) regionally sympatric (that is, Alpine monospecific populations) and (iii) locally sympatric (that is, Alpine localities containing both species within max. 300 m distance from one another). In addition, representatives of the diploid parents of the polyploids, D. fuchsii (19 individuals) and D. incarnata (23 individuals) were sampled from the same areas as the allotetraploid taxa (Supplementary Table S1). From all individuals, fresh flowers were collected in the field and immediately dried and stored in silica gel until DNA extraction following Hedrén et al. (2001).

The geographic location of the 24 polyploid Dactylorhiza populations studied here. Allopolyploid species are indicated by the colour of the symbols: red, D. majalis; green, D. traunsteineri; and violet, locally sympatric populations. The acronyms correspond to the localities given in Table 1.
Development and selection of microsatellite loci
Eight SSR loci (Table 2), newly reported here have been characterized from reference transcriptomes of the diploids D. incarnata and D. fuchsii (Paun et al. unpublished, available from http://www.botanik.univie.ac.at/systematik/projects/dactylorhiza/downloads.html). The SSR loci have been selected for length, polymorphism and to amplify in both diploid genomes, and primers were designed using QDD vers. 2 (Meglécz et al., 2010). We opted for EST-derived microsatellites as they are expected to be less prone to deletions resulting in null alleles (Ellis and Burke, 2007), a phenomenon otherwise particularly confounding in polyploid codominant data (Dufresne et al., 2014). As all SSR loci used here are (i) dinucleotide repeats, (ii) highly polymorphic (Table 2) and (iii) derived from total RNA, they are likely derived from intronic and UTR regions, and their variation is expected to be selectively (at least close to) neutral (Ellis and Burke, 2007). We initially also considered to use the SSR markers previously developed by Nordström and Hedrén (2007) from genomic DNA, but since these are all trinucleotide repeats showing relatively low levels of variability within and between populations (Hedrén et al., 2011), and were found unable to discriminate between the two sibling allotetraploids in Central Europe (Hedrén unpublished), these markers were not used in the present study.
To verify the power of discrimination of the SSR loci, we calculated for the 42 diploid individuals of D. fuchsii and D. incarnata the following indices: (i) the polymorphism information content (PIC) and observed heterozygosity (Ho) with MolKin v3.0 (Gutiérrez et al., 2005) and (ii) the probability of identity (PIdentity) of two genotypes by chance with GenAlEx v6.5 (Peakall and Smouse, 2012). Finally, as information derived from highly-polymorphic markers could be prone to misleading effects of homoplasy (that is, resulting in artificially decreased levels of genetic differentiation and false signals of introgression), we repeated the main genetic analyses using a reduced data set with the four least variable loci (D2, D8, D51 and D81) to check for consistency (Supplementary Figure S1).
DNA isolation and genotyping
Genomic DNA was extracted from every individual using the QIAGEN DNeasy Plant Mini Kit (Qiagen, Hilden, Germany). The SSR loci were first tested for uniplex amplification following Takayama et al. (2011), and then validated for PCR multiplexing in three different reactions in the following combinations: loci D8 and D81 (both labelled with FAM), D5, plus D16 and D55 (with PET), and D2, D51 and D52 (with VIC). Within each colour-coded combination, the alleles of the different loci could be identified and separated by size. For each multiplex PCR, we applied the 5′-tailed primer method (Boutin-Ganache et al., 2001) to label amplicons for visualization on the capillary sequencer. Forward primers were 5′-tailed with the 16-bp CAG sequence (CAGTCGGGCGTCATCA). We added GTTT 'pigtails' to the 5′ end of the reverse primer to facilitate addition of one adenosine nucleotide by Taq polymerase (Brownstein et al., 1996). We used the 2 × Type-It Multiplex PCR Master Mix (Qiagen), 0.3 μm of each forward primer, 1.5 μm of the labelled primer, 1.5 μm of each reverse primer and up to 50 ng of template DNA to a final volume of 5 μl. PCR amplifications were carried out in a GeneAmp PCR System 9700 thermocycler (Life Technologies, Darmstadt, Germany) using the following cycling conditions: initial denaturation at 95 °C for 5 min, 20 touchdown cycles at 95 °C for 30 s, 63 °C (decreased 0.5 °C per cycle) for 90 s and 72 °C for 30 s, then 20 cycles at 95 °C for 30 s, 53 °C for 90 s and 72 °C for 30 s, followed by a final elongation at 60 °C of 30 min. The fluorescence-labelled, Sephadex (GE Healthcare Bio-Sciences AB, Uppsala, Sweden)-filtered PCR products were separated on a capillary sequencer ABI 3130xl (Life Technologies) together with an internal size standard (GeneScan 600 LIZ v2.0, Life Technologies). Fragments were scored using the GeneMarker v. 2.4.0 software (SoftGenetics, State College, PA, USA), and they were further manually checked. To avoid any error bias in estimating allele dosage, the genotype of 38 polyploid individuals (10% of total) was confirmed by at least two independent repeats (starting from DNA extracts), revealing that the amplification and the scoring were fully reproducible.
To designate genotypic configurations for the tetraploid individuals, we used the microsatellite DNA allele counting-peak ratios method (Esselink et al., 2004). This method is based on quantitative values for microsatellite allele amplification peak areas and assesses the dosage of the amplified products (observed alleles) in the samples, by taking into account the potential for differential PCR amplification of alleles of different lengths. To investigate the reliability of this approach, we performed a Mantel test as implemented in the ecodist package (Goslee and Urban, 2007) in R version 3.1.1 (R Core Team, 2013) between two matrices of GST pairwise population measures calculated with the software Genodive v.2.0b26 (Meirmans and Van Tienderen, 2004). The first distance matrix was calculated using the quantitatively scored data with the microsatellite DNA allele counting-peak ratios method method. To calculate the second distance matrix, the allelic frequencies from a qualitative scoring were corrected using a maximum likelihood method modified from De Silva et al. (2005). The correlation between both matrices was highly significant (r=0.98; P<0.001), indicating a very good fit of the quantitative estimates.
Sibling species diversity and relatedness
To assess the genetic diversity for each population and species, the average number of alleles per locus (Num), the observed and the expected heterozygosity (Ho and, respectively, He), as well as the inbreeding coefficient (FIS=1- Ho / He) were computed with Genodive. Genetic diversity and rarity were estimated from allelic richness (A) and, respectively, the private alleles per locus (P), following a generalized rarefaction approach using ADZE v1.0 (Szpiech et al., 2008).
Although neopolyploids are expected to show disomic inheritance, a low rate of allele exchange between the two subgenomes each generation is sufficient to homogenize allele frequencies among the two subgenomes, even in fairly young allopolyploids (Meirmans and Van Tienderen, 2013). Allozyme studies in allotetraploid Dactylorhiza showed that intergenomic recombinations are rare, but existent (Hedrén, 1996; Aagaard et al., 2005). For three of the microsatellite loci investigated here (that is, D2, D52 and D55) for which a formal test is feasible due to non-overlapping parental allelic ranges (see Supplementary Figure S2), we observed a departure from disomic inheritance, based on G-tests of independent segregation of homoeologs (Aagaard et al., 2005; see Supplementary Table S2). In addition, computed inbreeding coefficients were close enough to zero to contradict a state of fixed heterozygosity at all eight loci (see below). Therefore, further analyses of genetic diversity and genetic structure assumed a model of tetrasomic inheritance.
Genetic structure and phylogeographical pattern
To visualize the genetic patterns among polyploids and the representatives of the diploid parental species, we constructed a Principal Coordinates Analysis (PCO), starting from a Bruvo pairwise individual distance (Bruvo et al., 2004) computed with the polysat package (Clark and Jasieniuk, 2011) in R. As they are free of underlying assumptions, multivariate analyses such as PCO are suitable to analyse mixed-ploidy data (Jombart et al., 2009; Dufresne et al., 2014). The Bruvo distance is based on the stepwise mutation model, taking allele length differences proportionally into consideration. It has the advantage that it can be used regardless of the ploidy of individuals and it is not affected by dosage uncertainty (Dufresne et al., 2014).
To investigate the population structure and search for potential signals of admixture, we used Structure ver. 2.3.3 (Pritchard et al., 2000) implemented on the Lifeportal of the University of Oslo (https://lifeportal.uio.no/). Two main analyses were performed with 25 million iterations each, plus 2.5 million iterations as burn-in. The first analysis included all 428 diploid and polyploid samples, coding the missing alleles in the diploids as recessive but not ambiguous alleles—according to the documentation for Structure version 2.3. This analysis used a model of uncorrelated allelic frequencies, as we expect the allelic frequencies to have changed independently in each of the two diploid species and the polyploids. The analysis was run starting with K=1 and finished at K=10, as no improvement in ∆K (Evanno et al., 2005) has been observed after K=2 (Supplementary Figure S3A). A second analysis was run with Structure for the 386 tetraploid individuals with K=1–30, and using correlated allele frequencies as the tetraploids compared here are young, sibling taxa.
As in the Structure analysis of all individuals a substructure was visible within D. incarnata, we performed two additional analyses (each with 5 million iterations plus 0.5 million as burn-in) to check whether this was still depicted when the polyploids were not included in the analyses. Therefore, one analysis included the 42 diploid individuals of D. fuchsii and D. incarnata, with K=1–10, and using uncorrelated allele frequencies, as the two diploid species are some of the most divergent species in the genus and their allelic frequencies are expected to change independently of each other. Finally, a separate analysis was done for the 23 individuals of D. incarnata, using K=1–10 and a model of correlated allele frequencies, as this was an analyses within a single species.
All Structure analyses used an admixture model and 20 independent runs for each K value. The results were summarized on the online platform Harvester (Earl and VonHoldt, 2011), which implements the Evanno method (Evanno et al., 2005) to assess the most likely K value given the data (Supplementary Figures S3 and S4). We produced combined files from the 20 replicates of the best K for each analysis using Clumpp v1.1.2 (Jakobsson and Rosenberg, 2007). The graphical representations of the Structure results were prepared with Distruct v1.1 (Rosenberg, 2004).
To test whether species and geographic groups within the polyploid group (that is, Pyrenees, Alps, Scandinavia and Britain) are biologically meaningful, we performed Analyses of Molecular Variance (AMOVA) as implemented in Genodive. Population subdivision was estimated from G-statistics analogues built from variance components obtained with AMOVA. Variance components and G-statistics were tested for significance using an exact non-parametric test and 10 000 permutations. In addition, based on geographic coordinates and pairwise GST (pwGST) values, we tested for isolation by distance (IBD) separately for each polyploid species, by using a Mantel test as implemented in the ecodist package (Goslee and Urban, 2007) in R. Further, we tested whether the IBD slope and intercept were significantly different with the Reduced Major Axis regression in the R-based lmodel2 package (Legendre, 2013). Exact significance tests were based on the Spearman correlation coefficients and 10 000 permutations.
Interspecific gene flow between sibling allopolyploids
To test for gene flow in sympatric tetraploid populations, we compared population pwGST measures (Nei, 1987) between the two species when found in allopatry, in regional sympatry and in local sympatry (see section Plant Sampling above) with a Kruskal–Wallis ANOVA. For this purpose, a post hoc non-parametric multiple comparison test (Siegel and Castellan, 1988) was performed at P=0.05 using the R package pgirmess (Giraudoux, 2013).
Furthermore, we used a log Bayes Factor approach with Migrate-n version 3.6.8 (Beerli and Palczewski, 2010) to estimate gene flow between the sibling allopolyploids in sympatry by comparing two models. First, we run a no-migration model with two populations (separating individuals of the two species from the Alps) with different effective population sizes (using a population estimator θ=4Neμ, where Ne is the effective population size and μ is the mutation rate). The second model was similar to the previous one but allowed different mutation-scaled migrations (Mi=mi/μ, with m1 the migration rate of D. majalis genes to D. traunsteineri and m2 the migration rate in the opposite direction). The calculation of log marginal likelihoods for the two models was done using the Bezier approximation, as this provides a better estimate of the marginal likelihood than the harmonic mean estimator (Beerli and Palczewski, 2010). These calculations relied on static heating with the temperatures of the four heated chains set at 1.0, 1.5, 3.0 and 100 000. Each hot and cold chain was run for 400 million generations per locus (20 replicates) with samples taken from the cold chain every 100 000 steps after a 100 000 burn-in per replicate. A random tree was used as a starting genealogy, and the initial theta and migration rates were estimated from FST calculations. We used slice sampling and uniform prior distribution to estimate θ (range=0–100, mean=50) and M (range=0–300, mean=150 and delta=250), respectively.
As Migrate-n is built for diploids (Beerli and Palczewski, 2010), we 'diploidized' our data choosing two random alleles per locus for each tetraploid individual. To assess the impact of this approach, five different data sets have been independently generated and run under both population models. The analyses used the Brownian approximation of the SMM and with mutation rate modifiers for each locus calculated according to the number of observed alleles. All analyses were run on the CIPRES Science Gateway (Miller et al., 2010).
Results
Microsatellite loci
Across the diploid individuals, the microsatellite loci showed between 7 and 29 alleles per locus, corresponding to a high level of polymorphism (PIC mean±s.e.: 0.8±0.1 and cumulative PIdentity 2.8 × 10−12, Table 2). As the number of alleles per individual is double for allotetraploids as compared with diploids (that is, larger effective population sizes) and the total numbers of alleles per locus amplified across all polyploid individuals were higher (between 14 and 38 alleles per locus, Table 2), we expect an even higher resolution power within the polyploid group, as compared with the diploids. The analyses using the data set reduced to the four least variable loci (Supplementary Figure S1) were highly congruent to the analyses of the full data set indicating a negligible impact of homoplasy in our inferences.
Genetic diversity
Estimates of genetic diversity for the studied tetraploid populations are presented in Table 1. On average, the microsatellite loci revealed 2.8 different alleles per locus per polyploid individual. The effective number of alleles per population across loci ranged from 2.0 (BRI4) to 7.5 (ALP13). In general, D. traunsteineri populations showed lower values than D. majalis populations for all diversity measurements. However, the highest values were found in the locally sympatric populations (in particular the population ALP9). The least diverse populations were the British D. traunsteineri populations (BRI1–BRI5) and the most western Alpine population sampled here (ALP11). The Scandinavian populations of each species showed intermediate diversity levels, whereas Pyrenean populations of D. majalis had higher diversity than their counterparts. The private alleles followed a similar pattern, but with exceptions. The Pyrenean populations of D. majalis and the northern-most Scandinavian population of D. traunsteineri (that is, SCA5) showed a remarkably high number of private alleles. The absolute values of the inbreeding coefficients (FIS) were usually very low. However, a tendency towards inbreeding (that is, positive and high FIS values) was detected in three populations (D. traunsteineri SCA4, ALP16 and the mixed population ALP13). In comparison with polyploids, the diploid species showed intermediate values of effective number of alleles (8.3 and 7.1 for D. fuchsii, and D. incarnata, respectively). The same pattern was observed for allelic richness, but not for private alleles (Table 1). In this case, D. fuchsii showed a high proportion of private alleles (0.8) but not D. incarnata (0.3), potentially indicative of higher gene flow with the polyploids of the latter. In addition, the difference in proportion of private alleles could relate to the generally higher diversity of D. fuchsii in comparison with D. incarnata (Table 1; Pillon et al., 2007). This renders many D. fuchsii individuals included here to be more dissimilar to the actual maternal parents of the polyploids than the representatives of the paternal, D. incarnata species.
After rarefaction correction, at the regional scale the Alpine populations showed the highest levels of allelic richness (16.0±2.2) and private alleles (1.8±0.5). Populations in the Pyrenees also showed high levels of allelic richness (14.0±1.6) and private alleles (1.7±0.8). Scandinavia had intermediate values in both genetic diversity and private alleles (13.4±1.8 and 0.7±0.2, respectively), whereas Britain was the most genetically depauperate region (8.9±0.8 and 0.3±0.1).
Low species differentiation and shallow genetic structure
The PCO (Figure 2) showed most tetraploid individuals to have an intermediate position between the two diploids, which had extreme positions along the first ordination axis, explaining 22.0% of the variation across the data. Several D. traunsteineri individuals, in particular those from populations ALP11 and ALP16, occupied positions close to the D. incarnata individuals. The second axis, containing 20.7% of the total variation, separated the D. traunsteineri individuals from Britain and some of the individuals from Scandinavia from the rest. In addition, we observed a geographical clustering within the two diploid clusters. For example, the D. incarnata individuals from Britain, Pyrenees and Scandinavia formed three groups, respectively, whereas the individuals from the Alps spread out to the extremes of the D . incarnata cluster.

PCO of microsatellite data of Dactylorhiza. Ordination based on the Bruvo distance matrix among 386 Dactylorhiza allopolyploids and 42 diploid progenitors. Individuals are labelled according to geographical provenance by different colours: blue, Alps; orange, Scandinavia; yellow, Pyrenees; and green; Britain. Species are indicated by shapes of the symbols: crosses, D. incarnata; crossed circles, D. fuchsii; triangles, D. majalis; and circles, D. traunsteineri.
For the best partition (K=2) when analysing the data set including all individuals with Structure (Supplementary Figure S3A), the D. traunsteineri individuals from Scandinavia, Britain and most of the individuals of ALP11, ALP13 and ALP16 formed a separate cluster, separated from all other polyploid individuals (that is, including D. traunsteineri from ALP8, ALP9 and ALP15, plus all D. majalis individuals except one individual from ALP13). With regard to the diploids, all D. fuchsii individuals were assigned to the ‘majalis’ cluster, whereas the D. incarnata individuals were split between the ‘traunsteineri’ cluster (that is, some of the D. incarnata individuals sympatric to D. traunsteineri populations ALP11, BRI5 and SCA4) and the ‘majalis’ cluster. However, the latter separation disappeared if only the diploids were analysed—this analysis separated the diploid species into different clusters according to the best partition of the data for K=2 (Supplementary Figure S4A). When only D. incarnata was analysed, the best partitioning was obtained at K=3 with clusters largely correlated with the geographic origin of the material (Supplementary Figure S4B).
When analysing only the tetraploid individuals separately, all individuals of D. majalis (with the exception of four admixed individuals of the sympatric population ALP13) were assigned to one genetic group (Figure 3) for the best data partition identified (K=3; Supplementary Figure S3B). A second genetic cluster comprised D. traunsteineri individuals from Britain and a third cluster comprised individuals of D. traunsteineri from the Alps (from ALP11, and in part ALP13 and ALP16). The Scandinavian populations of D. traunsteineri, both from mainland and from Gotland, appear as admixed between the two other genetic clusters of D. traunsteineri. Interestingly, the rest of the D. traunsteineri individuals from the Alps (ALP8, the rest of ALP13 and ALP16, and from the sympatric populations ALP9 and ALP15) remained assigned to the general ‘majalis’ cluster. Admixture signals were found in several individuals of both taxa, but the admixture was more common in two of the sympatric populations (ALP9 and ALP13). For two suboptimal partitions (K=9 and K=16, Supplementary Figure S3B), several populations were assigned as independent clusters (for example, PYR1, PYR3, SCA1, SCA2 and ALP4). Admixture appeared to affect more significantly the Alps region, but also populations from Yorkshire (BRI1 and BRI2, appearing as admixed between Scandinavian and Scottish populations, for K=9) and SCA5 (for K=16). Independent structure analyses on each of the three locally sympatric populations with a bound model of K=2 (Supplementary Figure S5) were unable to assign individuals to different clusters for ALP9 and ALP15. However, when analysing all tetraploid individuals together (Figure 3) for K=16 the D. majalis, and, respectively, the D. traunsteineri individuals sympatric in ALP9 (that is, the type locality of D. traunsteineri) are generally assigned to different genetic clusters.

Bayesian admixture analysis using structure on 386 allopolyploid Dactylorhiza individuals grouped to the optimal (K=3) and two best suboptimal (K=9 and 16) log-likelihood partitions. Each bar represents a different individual, whereas each segment length is proportional to the estimated membership of each group. The three locally sympatric populations are marked with a *.
The AMOVA (Table 3) indicated a high genetic similarity between the sibling polyploid species (GCT=0.02). However, a phylogeographical signal was present in the data, as the geographic origin accounted for a non-negligible amount of genetic variance (GCT=0.10), similar to the amount accounted for by grouping species by geographical origin (GCT=0.11). Congruent with the large geographical effect, D. traunsteineri exhibited significant IBD, as evidenced by the positive, significant correlation between pairwise geographic and genetic distances (Mantel’s r=0.42; P<0.001), as well as the Reduced Major Axis analysis (Supplementary Figure S6). However, D. majalis showed a positive, but non-significant IBD (r=0.24, P=0.237), despite the similar distance scale between its populations and those of D. traunsteineri.
Interspecific gene flow in sympatric populations
Although interspecies pwGST were mostly low (average±s.e.: 0.10±0.01), congruent with the AMOVA results, we found significant differences between the three categories with regard to the proximity of individuals of one species to the other: allopatric, regionally sympatric and locally sympatric (Kruskal–Wallis H=23.55, df=2, P<0.001). Congruent with the structure analyses, the Alpine populations showed high levels of gene flow between the sibling species as indicated by the significantly lower pwGST values than for the allopatric comparisons (Figure 4). Co-ocurrence of the two species at the same site (that is, local sympatry) showed the lowest values of pwGST (0.008±0.001). However, these values were not significantly different from the rest of the comparisons in the sympatric area (P>0.05 in post hoc multiple test). Finally, the allopatric pairwise comparisons between populations of the two allopolyploids showed significantly higher differentiation than comparisons at the level of the sympatric region (that is, Alps, P<0.05).

Boxplot of population pairwise genetic divergence (pwGST) between sibling Dactylorhiza allopolyploid populations when found in allopatry, in regional sympatry (i.e., in the Alps) and in local sympatry (i.e., within max. 300 m from one another). The first boxplot displays the overall variation in pwGST. Outliers are marked as circles. Medians not significantly different at P<0.05 are indicated by the same letter (non-parametric multiple comparison test).
The Migrate-n analyses revealed strong support for the model including gene flow between the sibling tetraploid species. The log-likelihood for the full model with gene flow and for the model without gene flow was −119203.06 and −3172370.18, respectively, which differed by log Bayes Factor of more than 6 millions. For the full model, the Bayesian inference indicated a high level of gene exchange between the species but migration rates did not significantly differ (gene flow from D. majalis to D. traunsteineri, Mmaj-trau=2.4–6.7 and gene flow from D. traunsteineri to D. majalis, Mtrau-maj=1.3–3.8). The average population sizes differed between species (θtrau=1.8–2.4 and θmaj=2.1–6.8) but their confidence intervals overlapped (Supplementary Table S3). Estimates of effective population sizes, migration rates and log-likelihood slightly varied between data sets, but were in general congruent (Supplementary Table S3).
Discussion
Genetic differentiation, multiple origins and phylogeographic pattern
By using highly variable microsatellites, this study corroborates the lack of genetic differentiation (GCT=0.02) between the sibling allopolyploids D. majalis and D. traunsteineri, previously observed with slower evolving makers (for example, Pillon et al., 2007). The 'porous' genetic boundaries identified do not clearly delimit the recognized taxonomic entities within the polyploids; the geography of populations alone explains a larger part of the genetic variance than the currently accepted species delimitation.
In spite of the remarkable absence of a clear genetic differentiation between the Dactylorhiza allopolyploids, our genetic analyses provided information about the phylogeographic history of the D. majalis complex. Our results generally confirm the genetic contiguity of D. majalis s.s. (Pillon et al., 2007), but point to a high intra-population diversity, most probably as a result of a relatively large effective population size and frequent outcrossing in this polyploid lineage. A latitudinal gradient is evident in allelic richness, with highest values in the Pyrenees and the Alps. This pattern is mirrored by measurements of the private alleles, indicating that the Pyrenees could have acted as a glacial refugial area for the D. majalis lineage, perhaps together with the previously recognized area around the Alps (Nordström and Hedrén, 2009). However, the lower diversity of the Scandinavian D. majalis populations is in agreement with a post-glaciation colonization of this area (Hewitt, 1999; Nordström and Hedrén, 2008).
In contrast, D. trausteineri showed higher heterogeneity—potentially due to more recent and polytopic origins of this taxon (Nordström and Hedrén, 2009), but less diverse populations compared with D. majalis. With the SSR data available, it is difficult to determine the exact number of origins for D. traunsteineri. However, the assignment analysis of structure revealed at least three different lineages within D. traunsteineri (two in the Alps and one in Britain), but it is unclear whether they are a product of independent origins, or rather only reflect IBD. The genetic patterns as observed in the PCO (Figure 2) and the structure results of the polyploid analysis (Figure 3) support a stepwise connectivity in the D. traunsteineri lineage from south towards north and north-west (that is, Alps-Scandinavia and Scandinavia-Britain), congruent with the two migration routes to Scandinavia identified by Nordström and Hedrén (2008). Furthermore, D. traunsteineri showed a more prominent IBD than D. majalis (see Supplementary Figure S6) despite the similar distance scales of the distribution of the two taxa. The difference in the IBD patterns between these allopolyploids cannot be explained by different pollinator behaviour (both are pollinated by food deception, mainly by bumblebees; Sletvold et al., 2010) or dispersal, given the similar tiny dust-like seeds (Arditti and Ghani, 2000). The distribution of terrestrial orchids is broadly driven by a general dispersal limitation to some metres from the parent plant (McCormick and Jacquemyn, 2014). However, occasional long-distance dispersal events by wind of up to 2000 km have been inferred (Arditti and Ghani, 2000; Jersáková and Malinová, 2007; Vandepitte et al., 2012). Long-distance, exceptional connections are also visible in the structure results for our data, for example, between Alps and Scandinavia (ALP13-SCA5) and between Scandinavian and Yorkshire populations (for K=9; Figure 3). Because of increased effective population sizes, genetic drift is less efficient in polyploids (Moody et al., 1993) and altogether drift does not appear to be a major evolutionary force in Orchidaceae (Phillips et al., 2012; Vanden Broeck et al., 2014). Multiple origins in different geographical areas and/or a disjunct distribution with strong geographical barriers are potential explanations for the increased IBD pattern observed within D. traunsteineri as compared with D. majalis.
Alternatively, the observed difference in IBD may relate to historical distribution patterns of these taxa. Dactylorhiza majalis is a lowland taxon, which before modern agriculture may have had a wide and relatively continuous distribution in the central European lowlands. The weak differentiation between regions we observed here may thus be explained by a high connectivity of historical populations (Dupouey et al., 2002; Lindborg and Eriksson 2004). In contrast, D. traunsteineri is predominantly distributed in upland areas in central Europe and it extends much farther to the north into boreal and low-mountain areas of Scandinavia. The regional populations of D. traunsteineri may thus have been isolated for longer, possibly since the early post-glacial period, and its populations may have had more time to diverge from each other.
Regarding 'D. ebudensis' (that is, here population BRI5) that has been recently embedded within D. traunsteineri (Bateman and Denholm, 2012), the microsatellites analysed here confirm its genetic similarity to other D. traunsteineri from Britain (Figure 3), but places it in an extreme position along the second axis in the PCO (Figure 2). The population of BRI5 is occupying a distinct habitat (young dune slacks on the island of North Uist) from the rest of the polyploid populations, and it is one of the largest metapopulations of Dactylorhiza polyploids in Britain. However, its effective number of alleles, allelic richness and private alleles are some of the lowest from the D. traunsteineri populations studied here, with values comparable to those of very small populations with 10 or less individuals (for example, ALP11 and BRI4). This supports a recent establishment of the population on this young dune system (hypothesized to be younger than 2500 years; Paun et al., 2010; Bateman, 2011), most probably as a result of long-distance dispersal from a D. traunsteineri source population elsewhere in Britain, and that it experienced a fairly strong bottleneck during establishment.
Gene flow among sibling allopolyploids
The area with the smallest genetic differentiation between D. majalis and D. trausnteineri was the Alps, where the two polyploids are sympatric. Here, lineage recombination among D. majalis and D. traunsteineri was revealed by extensive admixture. Remarkably, most Alpine D. traunsteineri populations, including its type locality near Schwarzsee (ALP9), were genetically similar to D. majalis populations from the Alps, but also to D. majalis from areas in which D. traunsteineri was absent. This points towards a largely unidirectional flow of D. majalis genes towards D. traunsteineri, probably due to swamping of D. traunsteineri as a result of larger effective population sizes of D. majalis, and due to larger inflorescences with more flowers in the latter. This is further supported by the Migrate-n results, even if the difference in rates of gene flow in the alternative directions was not found to be significant. The comparison between interspecific pairwise differentiation (Figure 4) between allopatric and, respectively, locally sympatric pairs of populations corroborated the extensive gene flow at local scale, but with visible effects at the scale of the entire Alps. This argument is supported also by the very weak pre-mating barriers in the genus (Aagaard et al., 2005), as Dactylorhiza species are food-deceptive and depend on a generalist pollinator spectrum (Ferdy et al., 2001; Cozzolino et al., 2004; Sletvold et al., 2010; Sletvold and Agren, 2011). The sibling allopolyploids investigated here show at least partly overlapping flowering time in sympatry (Bateman, 2011; personal observation) making gene flow possible. At the polyploid level, mechanisms such as homeologs repatterning could quickly achieve an increased postzygotic isolation between sister taxa (Oka, 1953; Lynch and Force, 2000; Gaeta and Pires, 2010); however, in some systems differential loss of duplicate gene copies is either rare or does not contribute significantly to postzygotic reproductive isolation (Modliszewski and Willis, 2012). In such cases, hybridization between sibling polyploids generally augment the level of genetic diversity in polyploids, like for example in Glycine tabacina (Doyle et al., 2004), in Mimulus sookensis (Modliszewski and Willis, 2012) and in autopolyploids Houstonia (Glenon and Church, 2015). The fairly high level of interspecific gene flow between D. majalis and D. traunsteineri suggests that any homeologs repatternings that has accumulated since the allopolyploidization events did not contribute significantly to postzygotic reproductive barriers in this group. Rather, the genomes of the two allopolyploids appear to be porous for introgression of genes from related polyploid genomes.
Gene flow between allopolyploids and their parents
Several previous studies have reported backcrossing of tetraploids with their diploid parental lineages on secondary contact in Dactylorhiza (for example, Hedrén, 2003; Aagaard et al., 2005; Nordström and Hedrén, 2008; De Hert et al., 2011, 2012), but also in other polyploid complexes (for example, Burton and Husband, 2001; Stift et al., 2010). This may be instrumented either through the production of unreduced gametes in the diploids or via a triploid generation of reduced fertility (De Hert et al., 2011). Our results support such an introgression, at least in some populations of D. traunteineri which could have hybridized in particular with D. incarnata. Indeed, the population ALP11 and most of the D. traunsteineri individuals from ALP16 and ALP13 appear in the PCO analysis (Figure 2) more related to the diploid D. incarnata than to the rest of the allotetraploids. This inference is not contradicted by the structure results on the data set including all individuals, which place for example the D. traunsteineri individuals from ALP11 and the sympatric D. incarnata individuals in the same genetic cluster. The D. traunsteineri population of ALP11 is a very small polyploid population (ca. 10 individuals), and sits in the middle of a large diploid D. incarnata stand. However, the D. traunsteineri individuals from ALP11 have been confirmed to be tetraploid by flow cytometry (Temsch and Paun, unpublished). We hypothesize that they may be the product of recent backcrossing of D. incarnata alleles into the polyploid background of D. traunsteineri, most likely via formation of unreduced gametes by the diploids. In general, introgressive gene flow between cytotypes after secondary contact has been reported more frequently in the direction from the diploid to the tetraploid level (for example, Ståhlberg, 2007). Although evidence for local hybridization and introgression between Dactylorhiza diploids and allotetraploids exists (Hedrén, 2003), postzygotic barriers have been demonstrated to prevent local breakdown of species boundaries as a result of genic incompatibilities and localized pollen flow, in combination with different microhabitat preference (De Hert et al., 2011, 2012). For example, crossing experiments of De Hert et al. (2012) document strong post-mating reproductive barriers that prevent local breakdown of species boundaries in mixed populations of D. incarnata, D. fuchsii and allopolyploid D. praetermissa, despite identifying frequent F1 hybrids in the natural populations. Although rare, introgression across ploidy barriers could however in time lead to significant alterations in the morphological, ecological and genetic features of polyploid species (Chapman and Abbot, 2010).
Recombination, maintenance of phenotypes and the fate of sibling allopolyploids
It has been hypothesized that gene flow between allopolyploid lineages, and between the polyploid and diploid level increases their genetic variability and widens the genetic basis for further adaptation at the polyploid level, enhancing the evolutionary success of allopolyploid species (Doyle et al., 1999; Petit et al., 1999; Meimberg et al., 2009). However, extensive gene flow between sibling polyploids of different ages may bring together different genetic materials into a common gene pool in sympatry. This genetic homogenization will have a significant effect on the subsequent evolutionary trajectories of the sibling polyploids, potentially leading to lineage swamping into an enriched panmictic species (Doyle et al., 2004; Soltis et al., 2010). Interestingly, despite the high level of gene flow in sympatry, the allopolyploids studied here maintain their phenotypic distinctiveness (Soó, 1980; Baumann and Künkele, 1988; Delforge, 2001). This phenomenon has been also reported for example in Asplenium allooctoploids (Perrie et al., 2010). In the context of the extensive gene flow and similar genomic background in Dactylorhiza sibling allopolyploids, we expect a fairly strong divergent selection to act on restricted genomic regions responsible for differential adaptation (Wu, 2001; Nosil et al., 2009) in order to maintain the observed phenotypic divergence.
Previous studies have consistently inferred the patterns of gene expression (Paun et al., 2011) and DNA methylation (Paun et al., 2010) among D. majalis and D. traunsteineri to have been shaped by divergent selection in response to different environmental pressures, in particular related to water availability and temperature differences in the native environments. Indeed, after iterative polyploidization events, phenotypic divergence between the resulting lineages can result from reciprocal silencing of homeologs (Lynch and Force, 2000; Bikard et al., 2009) and other divergent epigenetic alterations in response to the microenvironment (Bossdorf et al., 2008; Jablonka and Raz, 2009). The inheritance of epigenetic alterations in the face of gene flow could be maintained by mechanisms such as paramutation (Meyer et al., 1993; Alleman et al., 2006) or trans-chromosomal interactions, which have been recently demonstrated during Arabidopsis hybridization events (Greaves et al., 2013). Alternatively, the phenotypic divergence between sibling allopolyploids could be maintained by disruptive selection at a small number of loci (‘genomic islands’; Nosil et al., 2009; Renaut et al., 2013) within the otherwise porous genomic landscape. Genetic mechanisms such as divergence hitchhiking can allow ecological divergence in the presence of gene flow (Via, 2012; Feder et al., 2012). Ongoing investigations based on gene expression and genome-wide screen studies will help to discern between the proposed alternative hypotheses.
Data Archiving
Data accessibility Source data and R scripts used for this work are available from the Dryad Digital Repository (http://dx.doi.org/10.5061/dryad.9hb89). The separate worksheets refer to information related to (1) microsatellite data, (2) pairwise geographical and genetic distances of D. majalis, (3) pairwise geographical and genetic distances of D. traunsteineri, (4) population pairwise GST data and (5) additional parameters for analysis. The sequences of the microsatellite regions are available from GenBank, accession numbers KJ209801 to KJ209816.
Change history
16 January 2019
Since the publication of this article, the authors have noted that the input data in Fig. 3 is missing. The figure has been replaced with the correct version. The mistake does not affect the conclusions of the paper. We apologise for any inconvenience caused by this error.
References
Aagaard SMD, Såstad SM, Greilhuber J, Moen A . (2005). A secondary hybrid zone between diploid Dactylorhiza incarnata ssp. cruenta and allotetraploid D. lapponica (Orchidaceae). Heredity 94: 488–496.
Alleman M, Sidorenko L, McGinnis K, Seshadri V, Dorweiler JE, White J et al. (2006). An RNA-dependent RNA polymerase is required for paramutation in maize. Nature 442: 295–298.
Arditti J, Ghani A . (2000). Numerical and physical properties of orchid seeds and their biological implications. New Phytol 145: 367–421.
Bateman RM . (2011). Glacial progress: do we finally understand the narrow-leaved marsh-orchids? New J Bot 1: 2–15.
Bateman RM, Denholm I . (2012). Taxonomic reassessment of the British and Irish tetraploid marsh-orchids. New J Bot 2: 37–55.
Baumann H, Künkele S . (1988) Die Orchideen Europas. Kosmos Naturführer: Stuttgart.
Beerli P, Palczewski M . (2010). Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 185: 313–326.
Bikard D, Patel D, Le Metté C, Giorgi V, Camilleri C, Bennett MJ et al. (2009). Divergent evolution of duplicate genes leads to genetic incompatibilities within A. thaliana. Science 323: 623–626.
Bossdorf O, Richards CL, Pigliucci M . (2008). Epigenetics for ecologists. Ecol Lett 11: 106–115.
Boutin-Ganache I, Raposo M, Raymond M, Deschepper CF . (2001). M13-tailed primers improve the readability and usability of microsatellite analyses performed with two different allele-sizing methods. Biotechniques 31: 28.
Brownstein MJ, Carpten JD, Smith JR . (1996). Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques 20: 1004–1010.
Bruvo R, Michiels NK, D’Souza TG, Schulenburg H . (2004). A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol Ecol 13: 2101–2016.
Burton TL, Husband BC . (2001). Fecundity and offspring ploidy in matings among diploid, triploid and tetraploid Chamerion angustifolium (Onagraceae): consequences for tetraploid establishment. Heredity 87: 573–582.
Chapman MA, Abbott RJ . (2010). Introgression of fitness genes across a ploidy barrier. New Phytol 186: 63–71.
Chaudhary B, Flagel L, Stupar RM, Udall JA, Verma N, Springer NM, Wendel JF . (2009). Reciprocal silencing, transcriptional bias and functional divergence of homeologs in polyploid cotton (Gossypium. Genetics 182: 503–517.
Chester M, Gallagher JP, Symonds VV, Cruz da Silva AV, Mavrodiev EV, Leitch AR et al. (2012). Extensive chromosomal variation in a recently formed natural allopolyploid species, Tragopogon miscellus (Asteraceae). Proc Natl Acad Sci 109: 1176–1181.
Clark LV, Jasieniuk M . (2011). POLYSAT: an R package for polyploid microsatellite analysis. Mol Ecol Resour 11: 562–566.
Comai L, Madlung A, Josefsson C, Tyagi A . (2003). Do the different parental ‘heteromes’ cause genomic shock in newly formed allopolyploids? Philos Trans R Soc Lond B Biol Sci 358: 1149–1155.
Cozzolino S, D’Emerico S, Widmer A . (2004). Evidence for reproductive isolate selection in Mediterranean orchids: karyotype differences compensate for the lack of pollinator specificity. Proc R Soc B Biol Sci 271(Suppl): S259–S262.
Cozzolino S, Cafasso D, Pellegrino G, Musacchio A, Widmer A . (2003). Fine-scale phylogeographical analysis of Mediterranean Anacamptis palustris (Orchidaceae) populations based on chloroplast minisatellite and microsatellite variation. Mol Ecol 12: 2783–2792.
Cozzolino S, Widmer A . (2005). Orchid diversity: an evolutionary consequence of deception? Trends Ecol Evol 20: 487–494.
De Hert K, Jacquemyn H, Van Glabeke S, Roldán-Ruiz I, Vandepitte K, Leus L et al. (2011). Patterns of hybridization between diploid and derived allotetraploid species of Dactylorhiza (Orchidaceae) co-occurring in Belgium. Am J Bot 98: 946–955.
De Hert K, Jacquemyn H, Van Glabeke S, Roldán-Ruiz I, Vandepitte K, Leus L et al. (2012). Reproductive isolation and hybridization in sympatric populations of three Dactylorhiza species (Orchidaceae) with different ploidy levels. Ann Bot 109: 709–720.
De Silva HN, Hall AJ, Rikkerink E, McNeilage MA, Fraser LG . (2005). Estimation of allele frequencies in polyploids under certain patterns of inheritance. Heredity 95: 327–334.
Delforge P . (2001) Guide des Orchidées d´Europe, d´Afrique du Nord et du Proche-Orient. Delachaux et Niestlé: Lausanne & Paris.
Devos N, Raspé O, Oh S-H, Tyteca D, Jacquemart A-L . (2006). The evolution of Dactylorhiza (Orchidaceae) allotetraploid complex: Insights from nrDNA sequences and cpDNA PCR-RFLP data. Mol Phyl Evol 38: 767–778.
Doyle JJ, Doyle JL, Brown AHD . (1999). Origins, colonization, and lineage recombination in a widespread perennial soybean polyploid complex. Proc Natl Acad Sci USA 96: 10741–10745.
Doyle JJ, Doyle JL, Rauscher JT, Brown AHD . (2004). Evolution of the perennial soybean polyploid complex (Glycine subgenus Glycine: a study of contrasts. Biol J Linn Soc 82: 583–597.
Dufresne F, Stift M, Vergilino R, Mable BK . (2014). Recent progress and challenges in population genetics of polyploid organisms: an overview of current state-of-the-art molecular and statistical tools. Mol Ecol 23: 40–69.
Dupouey JL, Dambrine E, Laffite JD, Moares C . (2002). Irreversible impact of past land use on forest soils and biodiversity. Ecology 83: 2978–2984.
Earl DA, VonHoldt BM . (2011). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4: 359–361.
Ellis JR, Burke JM . (2007). EST-SSRs as a resource for population genetic analyses. Heredity 99: 125–132.
Esselink GD, Nybom H, Vosman B . (2004). Assignment of allelic configuration in polyploids using the MAC-PR (microsatellite DNA allele counting-peak ratios) method. Theor Appl Genet 109: 402–408.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14: 2611–2620.
Feder JL, Egan SP, Nosil P . (2012). The genomics of speciation-with-gene-flow. Trends Genet 28: 342–350.
Ferdy J, Loriot S, Sandmeier M, Lefranc M, Raquin C . (2001). Inbreeding depression in a rare deceptive orchid. Can J Bot 79: 1181–1188.
Gaeta RT, Pires JC . (2010). Homoeologous recombination in allopolyploids: the polyploid ratchet. New Phytol 186: 18–28.
Giraudoux P . (2013). pgirmess: Data analysis in Ecology. R package version 1.5.9. Available at http://CRAN.R-project.org/package=pgirmess.
Glennon KL, Church SA . (2015). Gene flow contributes to genetic diversity of tetraploid populations of the North American plant genus Houstonia (Rubiaceae). Rhodora 117: 41–66.
Goslee SC, Urban DL . (2007). The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw 22: 1–19.
Greaves IK, Groszmann M, Wang A, Peacock WJ, Dennis ES . (2013). Inheritance of trans chromosomal methylation patterns from Arabidopsis F1 hybrids. Proc Natl Acad Sci USA 111: 2017–2022.
Gutiérrez JP, Royo LJ, Alvarez I, Goyache F . (2005). MolKin v2.0: a computer program for genetic analysis of populations using molecular coancestry information. J Hered 96: 718–721.
Hedrén M . (1996). Genetic differentiation, polyploidization and hybridization in northern European Dactylorhiza (Orchidaceae): Evidence from allozyme markers. Plant Syst Evol 201: 31–55.
Hedrén M . (2003). Plastid DNA variation in the Dactylorhiza incarnata/maculata polyploid complex and the origin of allotetraploid D. sphagnicola (Orchidaceae). Mol Ecol 12: 2669–2680.
Hedrén M, Fay M, Chase M . (2001). Amplified fragment length polymorphisms (AFLP) reveal details of polyploid evolution in Dactylorhiza (Orchidaceae). Am J Bot 88: 1868–1880.
Hedrén M, Nordström S, Bateman RM . (2011). Plastid and nuclear DNA marker data support the recognition of four tetraploid marsh orchids (Dactylorhiza majalis sl, Orchidaceae) in Britain and Ireland, but require their recircumscription. Biol J Linn Soc 104: 107–128.
Hewitt GM . (1999). Post-glacial re-colonization of European biota. Biol J Linn Soc 68: 87–112.
Hultén E, Fries M . (1986) Atlas of North European Vascular Plants (North of the Tropic of Cancer) Vols. I–III, Koeltz Scientific Books: Königstein, Germany.
Jablonka E, Raz G . (2009). Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q Rev Biol 84: 131.
Jakobsson M, Rosenberg NA . (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806.
Jersáková J, Malinová T . (2007). Spatial aspects of seed dispersal and seedling recruitment in orchids. New Phytol 176: 237–241.
Jombart T, Pontier D, Dufour A . (2009). Genetic markers in the playground of multivariate analysis. Heredity 102: 330–341.
Kropf M, Renner SS . (2008). Pollinator-mediated selfing in two deceptive orchids and a review of pollinium tracking studies addressing geitonogamy. Oecologia 155: 497–508.
Kull T, Hutchings MJ . (2006). A comparative analysis of decline in the distribution ranges of orchid species in Estonia and the United Kingdom. Biol Cons 129: 31–39.
Legendre P . (2013). lmodel2: Model II Regression. R package version 1.7-2. Available at http://CRAN.R-project.org/package=lmodel2.
Levin DA . (2013). The timetable for allopolyploidy in flowering plants. Ann Bot 112: 1201–1208.
Lindborg R, Eriksson O . (2004). Historical landscape connectivity affects present plant species diversity. Ecology 85: 1840–1845.
Lynch M, Force A . (2000). The probability of duplicate gene preservation by subfunctionalization. Genetics 154: 459–473.
Madlung A, Wendel JF . (2013). Genetic and epigenetic aspects of polyploid evolution in plants. Cytogenet Genome Res 140: 270–285.
Marcussen T, Oxelman B, Skog A, Jakobsen KS. . (2010). Evolution of plant RNA polymerase IV/V genes: evidence of subneofunctionalization of duplicated NRPD2/NRPE2-like paralogs in Viola (Violaceae). BMC Evol Biol 10: 45.
McCormick M, Jacquemyn H . (2014). What constrains the distribution of orchid populations? New Phytol 202: 392–400.
Meglécz E, Costedoat C, Dubut V, Gilles A, Malausa T, Pech N et al. (2010). QDD: a user-friendly program to select microsatellite markers and design primers from large sequencing projects. Bioinformatics 26: 403–404.
Meimberg H, Rice KJ, Milan NF, Njoku CC, McKay JK . (2009). Multiple origins promote the ecological amplitude of allopolyploid Aegilops (Poaceae). Am J Bot 96: 1262–1273.
Meirmans PG, Van Tienderen PH . (2004). Genotype and Genodive: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4: 792–794.
Meirmans PG, Van Tienderen PH . (2013). The effects of inheritance in tetraploids on genetic diversity and population divergence. Heredity 110: 131–137.
Miller MA, Pfeiffer W, Schwartz T . (2010). Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE) 14 November 2010 New Orleans, LA, USA. pp 1–8 doi:10.1109/gce.2010.5676129.
Meyer P, Heidmann I, Niedenhof I . (1993). Differences in DNA methylation are associated with a paramutation phenomenon in transgenic petunia. Plant J 4: 89–100.
Modliszewski J, Willis JH . (2012). Allotetraploid Mimulus sookensis are highly interfertile despite independent origins. Mol Ecol 21: 5280–5298.
Moody ME, Mueller LD, Soltis DE . (1993). Genetic variation and random drift in autotetraploid populations. Genetics 134: 649–657.
Nei M . (1987) Molecular Evolutionary Genetics. Columbia University Press: New York, USA.
Nordström S, Hedrén M . (2007). Development of polymorphic nuclear microsatellite markers for polyploid and diploid members of the orchid genus Dactylorhiza. Mol Ecol Notes 7: 644–647.
Nordström S, Hedrén M . (2008). Genetic differentiation and post-glacial migration of the Dactylorhiza majalis ssp. traunsteineri/lapponica complex into Fennoscandia. Plant Syst Evol 276: 73–87.
Nordström S, Hedrén M . (2009). Evolution, phylogeography and taxonomy of allopolyploid Dactylorhiza (Orchidaceae) and its implications for conservation. Nord J Bot 27: 548–556.
Nosil P, Funk DJ, Ortiz-Barrientos D . (2009). Divergent selection and heterogeneous genomic divergence. Mol Ecol 18: 375–402.
Oka HI . (1953). Phylogenetic differentiation of the cultivated rice plant VI. The mechanism of sterility in the intervarietal hybrid of rice. Jpn J Breeding 2: 217–224.
Otto SP . (2007). The evolutionary consequences of polyploidy. Cell 131: 452–462.
Paun O, Bateman RM, Fay MF, Hedrén M, Civeyrel L, Chase MW . (2010). Stable epigenetic effects impact adaptation in allopolyploid orchids (Dactylorhiza: Orchidaceae). Mol Biol Evol 27: 2465–2473.
Paun O, Bateman RM, Fay MF, Luna JA, Moat J, Hedrén M et al. (2011). Altered gene expression and ecological divergence in sibling allopolyploids of Dactylorhiza (Orchidaceae). BMC Evol Biol 11: 113.
Paun O, Fay MF, Soltis DE, Chase MW . (2007). Genetic and epigenetic alterations after hybridization and genome doubling. Taxon 56: 649–656.
Peakall R, Smouse PE . (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research–-an update. Bioinformatics 28: 2537–2539.
Perrie LR, Shepherd LD, De Lange PJ, Brownsey PJ . (2010). Parallel polyploid speciation: distinct sympatric gene-pools of recurrently derived allo-octoploid Asplenium ferns. Mol Ecol 19: 2916–2932.
Petit C, Bretagnolle F, Felber F . (1999). Evolutionary consequences of diploid-polyploid hybrid zones in wild species. Trends Ecol Evol 14: 306–311.
Phillips RD, Dixon KW, Peakall R . (2012). Low population genetic differentiation in the Orchidaceae: implications for the diversification of the family. Mol Ecol 21: 5208–5220.
Pillon Y, Fay M, Hedrén M, Bateman R . (2007). Evolution and temporal diversification of western European polyploid species complexes in Dactylorhiza (Orchidaceae). Taxon 56: 1185–1208.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
R Core Team. (2013) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria. Available at https://www.R-project.org/.
Renaut S, Grassa CJ, Yeaman S, Moyers BT, Lai Z, Kane NC et al. (2013). Genomic islands of divergence are not affected by geography of speciation in sunflowers. Nat Commun 4: 1827.
Rosenberg NA . (2004). Distruct: a program for the graphical display of population structure. Mol Ecol Notes 4: 137–138.
Shaked H, Kashkush K, Ozkan H, Feldman M, Levy AA . (2001). Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell 13: 1749–1759.
Siegel S, Castellan JNJ . (1988) Nonparametric Statistics for the Behavioral Sciences. McGraw Hill: NY, USA.
Sletvold N, Grindeland JM, Agren J . (2010). Pollinator-mediated selection on floral display, spur length and flowering phenology in the deceptive orchid Dactylorhiza lapponica. New Phytol 188: 385–392.
Sletvold N, Agren J . (2011). Nonadditive effects of floral display and spur length on reproductive success in a deceptive orchid. Ecology 92: 2167–2174.
Soltis DE, Soltis PS, Pires JC, Kovarik A, Tate J . (2004). Recent and recurrent polyploidy in Tragopogon (Asteraceae): genetics, genomic, and cytogenetic comparisons. Biol J Linn Soc 82: 485–501.
Soltis DE, Buggs RJA, Doyle JJ, Soltis PS . (2010). What we still don’t know about polyploidy. Taxon 59: 1387–1403.
Soltis PS, Soltis DE . (2009). The role of hybridization in plant speciation. Annu Rev Plant Biol 60: 561–588.
Soó R de . (1980) Dactylorhiza. In: Tutin TG et al (eds). Flora Europea, Vol 5, Cambridge University Press: Cambridge, UK. pp 333–337.
Squirrel J, Hollingsworth PM, Bateman RM, Dickson JH, Light MHS, MacConaill M, Tebbitt MC . (2001). Partitioning and diversity of nuclear and organelle markers in native and introduced populations of Epipactis helleborine (Orchidaceae). Am J Bot 88: 1409–1418.
Ståhlberg D . (2007). Habitat differentiation, hybridization and gene flow patterns in mixed populations of diploid and autotetraploid Dactylorhiza maculata s.l. (Orchidaceae). Evol Ecol 23: 295–328.
Stift M, Bregman R, Oostermeijer JGB, van Tienderen PH . (2010). Other tetraploid species and conspecific diploids as sources of genetic variation for an autotetraploid. Am J Bot 97: 1858–1866.
Szpiech Z a, Jakobsson M, Rosenberg NA . (2008). ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24: 2498–2504.
Takayama K, S PL, König C, Kohl G, Novak J, Stuessy TF . (2011). A simple and cost-effective approach for microsatellite isolation in non-model plant species using small-scale 454 pyrosequencing. Taxon 60: 1442–1449.
Vanden Broeck A, Van Landuyt W, Cox K, De Bruyn L, Gyselings R, Oostermeijer G et al. (2014). High levels of effective long-distance dispersal may blur ecotypic divergence in a rare terrestrial orchid. BMC Ecol 14: 20.
Vandepitte K, Gristina AS, De Hert K, Meekers T, Roldán-Ruiz I, Honnay O . (2012). Recolonization after habitat restoration leads to decreased genetic variation in populations of a terrestrial orchid. Mol Ecol 21: 4206–4215.
Via S . (2012). Divergence hitchhiking and the spread of genomic isolation during ecological speciation-with-gene-flow. Philos Trans R Soc Lond B Biol Sci 367: 451–460.
Wu CI . (2001). The genic view of the process of speciation. J Evol Biol 14: 861–865.
Acknowledgements
We are grateful to Richard Bateman, Mark Chase, Laure Civeyrel, Mike Fay, Javier Luna, Ortrun Mittelsten Scheid, Peter Schönswetter, Laura Sedman, Andreas Tribsch, Franz Weberndorfer, Barbara Mable and three anonymous reviewers for their contribution to the work presented here. Natural England and the Forestry Commission (UK), and regional county administrations in Sweden are acknowledged for issuing necessary collecting permits. Sequencing the reference transcriptomes for microsatellite development has been done at the CSF Vienna (http://ngs.csf.ac.at). This study was funded by the Austrian Science Fund (FWF; projects P22260 and Y661 awarded to O Paun) and a Marie Curie IntraEuropean fellowship (TransAdaptation, awarded to F Balao).
Author contributions
OP designed the research and developed the microsatellite loci. FB, MH and OP collected the samples. MT and MTLR collected the data. FB performed most of the analyses. FB and OP wrote the paper. All authors improved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Balao, F., Tannhäuser, M., Lorenzo, M. et al. Genetic differentiation and admixture between sibling allopolyploids in the Dactylorhiza majalis complex. Heredity 116, 351–361 (2016). https://doi.org/10.1038/hdy.2015.98
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2015.98
This article is cited by
-
Dactylorhiza maculata agg. (Orchidaceae) in Central Europe: Intricate Patterns in Morphological Variability, Cytotype Diversity and Ecology Support the Single-Species Concept
Folia Geobotanica (2023)
-
Systematic reappraisal of marsh-orchids native to Scotland
Kew Bulletin (2023)
-
Systematics and conservation of British and Irish orchids: a "state of the union" assessment to accompany Atlas 2020
Kew Bulletin (2022)
-
Patterns of morpho-phenological and genetic variation of Brachypodium distachyon (L.) P.Beauv. complex in Tunisia
Genetic Resources and Crop Evolution (2022)
-
Morphological variability, cytotype diversity, and cytogeography of populations traditionally called Dactylorhiza fuchsii in Central Europe
Plant Systematics and Evolution (2021)
