
Abstract
The processes that have shaped the extraordinary species diversity in neotropical rainforests are poorly understood, and knowledge about the patterns of genetic diversity across species' ranges is scarce, in contrast to other regions of the globe. We have conducted a range-wide study of genetic diversity in a plant endemic to the Brazilian Atlantic Rainforest, Vriesea gigantea (Bromeliaceae), based on a combined data set of nuclear microsatellites and chloroplast (cp) DNA markers typed in 429 plants from 13 populations. The results indicate a strong negative correlation between genetic diversity and population latitude, consistent with historical forest expansion from the northern half of the present distribution range. A deep phylogeographic split exists between the Brazilian states of São Paulo and Rio de Janeiro at ca. 23 °S latitude, probably reflecting past population isolation within more than one glacial refuge during the climatic changes of the Pleistocene. A comparison of genetic structures at cpDNA and nuclear markers revealed a pollen/seed flow ratio of more than 3:1, thus indicating an important role of the pollinating animals (that is, bats) in shaping the population genetic structure of this species. Diversity was reduced for cpDNA markers in the island populations off the coast, and reduced diversity and increased differentiation were observed for both nuclear and cpDNA at the edges of the species' range. The link between patterns of genetic and species diversity supports the hypothesis that both were shaped by the same biogeographic processes, triggered by the climatic oscillations of the Pleistocene.
Similar content being viewed by others

Introduction
South America represents the most biodiverse subcontinent on Earth, where five of the world's biodiversity ‘hotspots’ are located (Myers et al., 2000). Despite an increasing interest in the evolutionary genetics of plant diversification in the neotropics (Rull, 2006; Barbará et al., 2008; Widmer et al., 2009), surprisingly little background information is available on the phylogeographic structure and patterns of gene flow across species' ranges in these ecosystems, compared with the temperate regions of the world (Hewitt, 2004). Biogeographic studies of taxa from the Amazon (Aleixo, 2004), the Andes (Pennington et al., 2002; Speranza et al., 2007) and the Brazilian Atlantic Rainforest (da Silva et al., 2004; Carnaval and Moritz, 2008) indicate the evolutionary complexity of the Quaternary history of South America, with a prominent role of range contractions and expansions due to the climatic changes of the Pleistocene.
Studies addressing the evolutionary history of the species-rich Southern part of the Brazilian Atlantic Rainforest taxa from a molecular phylogeographic perspective are extremely rare and include few studies of animals (Grazziotin et al., 2006; Cabanne et al., 2007, 2008; Tchaicka et al., 2007) and plants (for example, Lorenz-Lemke et al., 2005; Ledru et al., 2007). These studies provide first insights into the effects of historical forest expansions and contractions on present-day patterns of intraspecific variation, suggesting demographic responses to Pleistocene climate-induced changes in forest cover with a conspicuous north–south phylogeographic split for several taxa.
Bromeliaceae is an example of a large and well-described adaptive radiation in the neotropics including approximately 3000 species (Benzing, 2000). Systematic relationships in the family are relatively well understood compared with most other neotropical plant families, and bromeliads are increasingly being studied by population geneticists interested in microevolution and speciation in the neotropics (for example, Sarthou et al., 2001; Cavallari et al., 2006; Barbará et al., 2007, 2008). Bromeliads also show striking variation in breeding systems (from predominant inbreeding to predominant outcrossing), growth habits (epiphytic, terrestrial or saxicolous) and distributions (continuous forest species vs patchily distributed, rock-adapted ‘inselberg’ species).
Vriesea gigantea Gaud. is a perennial, diploid (2n=50 chromosomes, Palma-Silva et al., 2008) bromeliad species endemic to the Brazilian Atlantic Rainforest. It is epiphytic, saxicolous or terrestrial, occurring in the lowland Atlantic Rainforest at altitudes of up to 500 m. Its distribution range spans ca. 1600 km between the Brazilian states of Rio Grande do Sul (RS) and Espírito Santo (ES) (Figure 1; Smith and Downs, 1977). The species has a mixed-mating system (Paggi et al., 2007) with bat pollination (Sazima et al., 1999) and wind-based seed dispersal. Its populations are generally large and composed of patches of different sizes and densities. V. gigantea has an important role in its ecosystem as its rosettes form tank-like structures able to hold large amounts of water, thus supporting a vast number of associated animal species (Benzing, 2000). Recently, we have started to use species of the closely related genera, Vriesea and Alcantarea, for studies of the genetic underpinnings of adaptive radiation in the Tillandsioideae subfamily of Bromeliaceae. These two genera have diversified into multiple species adapted to life in continuous forest or on granitic ‘inselberg’ rock outcrops in the South American Atlantic Rainforest (Barbará et al., 2007, 2008; Paggi et al., 2007; Palma-Silva et al., 2007). This study provides vital genetic and phylogeographic data to inform ongoing studies on the genetics of population divergence and speciation in this group.

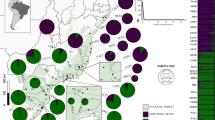
Map showing the current geographic distribution of Vriesea gigantea, including patterns of chloroplast (cp) and nuclear DNA diversity. (a) Populations sampled, pie charts representing the frequencies of cpDNA haplotypes. (b) Median-joining network of cpDNA haplotypes. Filled circles indicate the haplotypes, the size of each circle being proportional to the observed frequency of each type. The number of mutations required to explain transitions among haplotypes is indicated along the lines connecting the haplotypes.
We report and discuss range-wide patterns of nuclear and cpDNA diversity in V. gigantea. Specifically, we use our combined nuclear and organellar data to address the following questions: (1) Is there a north–south phylogeographic split in populations of this bromeliad species, as observed for several animal taxa previously? (2) If yes, what is the extent of genetic differentiation among southern and northern populations of the current distribution range and which past demographic processes may have shaped the phylogeographic patterns? (3) What is the relative contribution of seed versus pollen flow to historical gene flow, and what are the consequences of the observed dispersal patterns for population connectivity? We discuss phylogeographic and genetic structure in the light of available knowledge on the biogeographic history of the Brazilian Atlantic Rainforest and comment on implications for future work.
Materials and methods
Population sampling and DNA extraction
The populations were sampled from 2004 to 2006 from 13 forest fragments distributed across the Atlantic Rainforest (Table 1; Figure 1). The Atlantic Rainforest in southern and southeastern Brazil today is a mosaic of few large fragments of forest and a large number of small and disturbed fragments with crucial importance to the connectivity and persistence of the forest (Zaú, 1998). The majority of the sampled forest fragments were connected with other small fragments, exceptions being those fragments located in islands and in the southern part of the species distribution (see Table 1 and Figure 1). Individuals were sampled at least 5 m apart to avoid sampling close relatives. Population sampling was performed in such a way as to represent the complete geographical range of the species. Geographical distances between populations ranged from 34 km (FLSC-VBSC) to ca. 1600 km (TMRS-SLES). Fresh leaves (∼5 cm2) were collected and stored in liquid nitrogen or in 2% cetyltrimethyl ammonium bromide (CTAB) buffer until DNA extraction. Total genomic DNA was extracted as described by Palma-Silva et al. (2007).
Nuclear microsatellite analysis
A total of 10 nuclear microsatellite loci previously characterized for the bromeliad genera, Tillandsia and Guzmania, (Boneh et al., 2003) and for the species, Alcantarea imperialis and V. gigantea, (Palma-Silva et al., 2007) were selected for population genetic analysis, using fluorescent-labeled primers (6-FAM or JOE) and protocols as detailed in Palma-Silva et al. (2007).
Identification and characterization of chloroplast DNA (cpDNA) markers
The initial screening of polymorphism conducted with 21 regions, totaling ∼5,500 base pairs of the chloroplast genome of V. gigantea, yielded four polymorphic cpDNA microsatellite or simple sequence repeat loci (cpSSRs) and one single nucleotide polymorphism (cpSNP). For the four cpSSRs, primer pairs were designed from the regions sequenced using universal primers described by Taberlet et al. (1991), Sang et al. (1997) and Hwang et al. (2000). PCR was carried out in a total volume of 20 μl containing 10 ng DNA template, 1 × Bioline Taq buffer, 2 mM Bioline MgCl2, 100 μM deoxynucleotide triphosphate, 20 pmol forward primer, 20 pmol reverse primer and 2U Taq polymerase (Bioline, London, UK). Reactions were performed in a PE 9700 thermocycler (PE Applied Biosystems, Foster City, CA, USA) by using a standard cycling program: 95 °C for 3 min, 38 cycles at 94 °C for 30 s, 53 °C for 30 s, 72 °C for 30 s and a final elongation step at 72 °C for 10 min. The products were purified (QIAquick, West Sussex, UK) and sequenced using the BigDye terminator V3.1 cycle sequencing kit (Applied Biosystems) and a 3700 DNA Analyzer (Applied Biosystems). Forward and reverse sequences were aligned in SEQUENCHER version 4.1.2 (Gene Codes Corp., Ann Arbor, MI, USA). The characteristics and sequences of the primers designed for cpSSR loci are reported in Supplementary material 1. The primers were designed using the Primer3 programme (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). The cpSNP was identified by sequencing the chloroplast region rpoB using universal primers described on the KEW RBG website (http://www.kew.org/barcoding/protocols.html), using the same protocol described above for the remaining regions.
The chloroplast DNA (cpDNA) marker analysis
The cpSNP was amplified and sequenced for all samples as described above. For cpSSRs, all PCR amplifications were performed in a PE 9700 thermocycler (PE Applied Biosystems) in 10 μl reactions containing 10 ng DNA template, 1 × Bioline Taq buffer, 1.5 mM Bioline MgCl2, 100 μM deoxynucleotide triphosphate, 10 pmol either 6-FAM- or JOE-labeled forward primers, 10 pmol reverse primer and 0.5 U Taq polymerase (Bioline). A standard cycling program was used: 95 °C for 3 min, 30 cycles at 94 °C for 30 s, 58 °C for 30 s, 72 °C for 30 s and a final elongation step at 72 °C for 10 min. Microsatellite variants were resolved on a 3100 DNA analyzer (Applied Biosystems) and were precisely sized against ROX (Applied Biosystems) molecular size standard using GeneScan 3.7 and Genotyper 3.7 software (Applied Biosystems). The molecular basis of fragment length variation was confirmed by sequencing as described above.
Statistical analysis
The levels of genetic diversity within populations were evaluated by calculating the following statistics: (1) percentage of polymorphic loci (P); (2) number of private alleles (Apr); (3) allelic richness (Rs); (El Mousadik and Petit, 1996), which is a measurement of the number of alleles per locus standardized for sample size by rarefaction; (4) variance in allele size (Var), which reflects historical population size; (5) observed heterozygosity (HO); and (6) expected heterozygosity (HE) under Hardy–Weinberg equilibrium (Nei, 1978). All genetic diversity parameters were measured with the programs, Microsatellite Analyser (Dieringer and Schlötterer, 2003) and FSTAT 1.2 (Goudet, 1995). The existence of a latitudinal gradient in genetic diversity was tested using both linear regression and Spearman's rank correlation between diversity (allelic richness, variance in allele size and gene diversity) and population latitudes. The marginal populations (TMRS and SLES) were excluded from this analysis on the basis of their disproportionately great differentiation from the remaining populations (see the ‘Results’ section). The GENEPOP web version 3.5 (Raymond and Rousset, 1995) was used to estimate the inbreeding coefficient FIS (Weir and Cockerham, 1984).
The hypothesis that populations are differentiated because of isolation-by-distance (Wright, 1965) was tested by calculating the correlation between geographic and genetic distance matrices with a standardized Mantel test (Sokal and Rohlf, 1995) using FSTAT (Goudet, 1995). The significance was assessed through a randomization test using 10 000 Monte Carlo simulations. Although all populations were analyzed together in the Mantel test, comparisons that either included or excluded the two marginal populations were labeled as such in the resulting scatter plot.
F-statistics were estimated to quantify levels of genetic diversity within and among populations and to infer the degree of population subdivision. They were computed according to Weir and Cockerham (1984) by using the Microsatellite Analyser software (Dieringer and Schlötterer, 2003). The significance of each F-statistic was tested by resampling with 10 000 permutations. Bayesian clustering with the software program, STRUCTURE 2.0 (Pritchard et al., 2000), was used to assign individuals to genetic clusters (K) and to estimate admixture proportions (Q) for each individual. The proportion of membership for each cluster was calculated without the consideration of sampling localities. The analyses were carried out under the admixture model assuming independent allele frequencies and using a burn-in period of 50 000, run lengths of 300 000 and 10 iterations per K ranging from 1 to 13 to confirm stabilization of the summary statistics (Pritchard et al., 2000). To determine the most likely number of clusters (K) present in the data, we used the method proposed by Evanno et al. (2005), which is based upon an ad hoc measure ΔK that evaluates the second-order rate of change of the likelihood function with respect to K.
The cpSSR length and cpSNP variation were combined to define the haplotype of each individual. In addition, each population was characterized for levels of diversity at the cpDNA markers using: (1) the number of haplotypes detected in each sample, (2) HE—gene diversity (Nei, 1978) and (3) allelic richness (El Mousadik and Petit, 1996), estimated using the program CONTRIB (Petit et al., 1998). A median-joining haplotype NETWORK (Bandelt et al., 1999) was constructed based on cpDNA haplotypes using the program NETWORK 4.1.1.1. (http://www.fluxus-engineering.com). Initial runs using all the haplotype information yielded a complex structure. Hence, rapidly mutating characters were downweighted in subsequent runs following the user manual. For all other parameters, the default settings were used.
The analysis of molecular variance in the software ARLEQUIN 3.01 (Excoffier et al., 2005) was used to estimate the Φ-statistics (Weir and Cockerham, 1984) for nuclear microsatellite and cpDNA markers, hence allowing us to disentangle the effects of pollen and seed dispersal on the patterns of genetic variability. To estimate the relative contribution of pollen versus seed flow to total gene flow, the following formula from Ennos (1994) was applied:

where ΦSC(B) and ΦSC(M) are levels of population differentiation calculated from biparentally (nuclear) and maternally (cpDNA) inherited markers, respectively. The calculation of the pollen–seed flow ratio presented here is based on the assumption that maternal inheritance of cpDNA is the rule in V. gigantea as in most other angiosperms (Ennos et al., 1999).
Results
Nuclear microsatellite loci
All 10 microsatellite loci were polymorphic in V. gigantea, with an average of 16 alleles and ranging from 3 (Ai4.10) to 25 (VgF01) alleles per locus (Table 2). The observed and expected heterozygosity of the loci, estimated over all populations, ranged from 0.223 to 0.547 and from 0.354 to 0.715, respectively (Table 2). All loci departed significantly from Hardy-Weinberg equilibrium within populations at the 0.05 level (Table 2). A total of 40 alleles out of 160 were exclusive to a single population (=private alleles). Most private alleles were found in populations JASP, BESP, PTRJ, RIRJ and SLES. The populations JASP and SLES showed the highest number of private alleles (Table 3).
Genetic diversity in V. gigantea
Allelic richness (Rs) in populations of V. gigantea averaged over loci was 2.83 and ranged from 1.18 to 3.49, whereas the variance in allele size (Var) ranged from 1.20 to 82.71 with an average of 31.85. The observed and expected heterozygosity per population varied from 0.039 to 0.614 and 0.065 to 0.727, respectively. The inbreeding coefficients (FIS) were high and significant in most populations, as expected for this species, which is known to have high selfing rates (Paggi et al., 2007). A marked decrease in nuclear diversity was observed for the two marginal populations, TMRS and SLES (Table 3; Figure 2). Several loci were even fixed for a single allele there (population TMRS: loci Ai4.3, Ai4.11, Ai5.18, VgA04, VgF01, VgG02; population SLES: loci CT5, Ai4.10, Ai4.11). Except for locus Ai4.11, different loci were fixed for different alleles in opposite margins of the species range. The distribution of genetic diversity (variation in allele size, allelic richness and gene diversity) across the remaining populations was correlated with latitude (Supplementary Figure 1). Qualitative inspection of the data indicated that the center of nuclear genetic diversity for V. gigantea is represented by populations JASP, BESP, PTRJ and RIRJ, and diversity decreased steadily toward the south (Table 3).

Relationship between genetic divergence, based on FST, and geographical distances in kilometers among populations of Vriesea gigantea (Mantel test correlation: r=0.53; P<0.001). Pair-wise comparisons, including the marginal populations, TMRS and SLES, are indicated by closed circles, and all remaining population pairs are indicated by open circles.
Patterns of population divergence
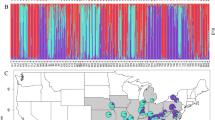
Significant differentiation among populations was indicated by FST of 0.211 (Table 2). Further, a significant Mantel correlation was observed between genetic distance (FST/(1−FST) and geographical distance (r2=0.53, P<0.001), suggesting the presence of isolation by distance (Figure 2). Pairs of populations involving the two marginal populations, TMRS and SLES, were more strongly differentiated than the remaining population pairs at all geographic distances (Figure 2). Bayesian analysis with STRUCTURE consistently identified K=6 clusters as shown in Figure 3. The admixture proportions (Q) for each individual plant are shown in Figure 4. The marginal populations, TMRS and SLES, were clustered separately from central populations, indicating restricted gene flow between central and marginal populations.

Magnitude of ΔK from structure analysis as a function of K (mean±s.d. over 10 replicates), calculated following the ΔK method proposed by Evanno et al. (2005), for Vriesea gigantea microsatellite data. The modal value of these distributions indicates the true K or the uppermost level of structure, here six ‘genetic clusters’.

Bayesian admixture proportions (Q) of individual plants of Vriesea gigantea for a K=6 population model. The K=6 ‘genetic clusters’, identified by the program STRUCTURE, are indicated in different colors. For population abbreviations, see ‘Materials and methods’. Brazilian Federal States: RS, Rio Grande do Sul; SC, Santa Catarina; PR, Paraná; SP, São Paulo; RJ, Rio de Janeiro; ES, Espírito Santo.
Chloroplast (cp) DNA haplotype diversity and distribution
Characterization of the cpDNA markers in populations of V. gigantea yielded two to five alleles per polymorphic site (Supplementary Table 1). One cpSNP was found, namely a transversion (T–C). The combined genetic information from the five polymorphic sites resulted in 13 distinct cpDNA haplotypes (H1–H13) (Tables 3 and 4; Figure 1), four of which were singletons. All populations were polymorphic, showing two to five haplotypes each, with the exception of population PTRJ (Tables 3 and 4; Figure 1), which showed a single fixed haplotype (H13). Populations JASP and BESP showed the highest number of haplotypes (five in each population) and values of allelic richness (2.349 and 2.810, respectively). Among the five populations with the lowest haplotype diversity were the two marginal populations (TMRS and SLES). The majority of populations showed moderate levels of gene diversity (HE) with a mean of HE=0.486 (Table 3), exceptions being the population of PTRJ (HE=zero) and the marginal populations (TMRS and SLES; HE=0.133 and 0.118, respectively) (Table 3).
Chloroplast (cp) DNA haplotype network
Median-joining analysis resulted in a haplotype network with two major haplogroups, one containing primarily haplotypes typical of the southern populations (from TMRS to BESP), and the other containing mainly haplotypes typical of the northern populations (from PTRJ to SLES) (Figure 1). These two major groups were separated by three mutational steps (Figure 1b). Moreover, haplotypes from the northern populations showed a high degree of geographic structure, with no haplotype found in more than one population (Table 4), and a higher number of mutational steps between haplotypes compared with the southern region (Figure 1b). In contrast, in the Southern group several haplotypes were shared among populations (Table 4). The haplotype network also indicates relatively recent population expansion in the southern group, where several haplotypes are connected by short branches with potentially ancestral, widespread haplotypes (H8 and H12).
Pollen versus seed flow
Using the values of genetic variance (analysis of molecular variance, Table 5) among populations for cpDNA markers (ΦSC=0.59) and for nuclear markers (ΦSC=0.21), the ratio of pollen flow to seed flow (Ennos, 1994) was estimated as ∼3.3, indicating that gene flow through pollen in V. gigantea is ca. threefold more efficient than through seeds.
Discussion
The genetics of population divergence and speciation represents one of the most hotly debated topics in evolutionary genetics (Coyne and Orr, 2004; Butlin and Ritchie, 2009), but knowledge on this topic is surprisingly scarce for continental adaptive radiations in the world's biodiversity hotspots such as those found in the neotropics (Rull, 2008). Bromeliaceae (bromeliads) have been identified as valuable models for studying the ecology and genetics of plant diversification (Benzing, 2000; Barbará et al., 2007, 2008; Wendt et al., 2008), but the necessary genetic and phylogeographic data required for interpreting divergence processes are largely missing. Range-wide genetic and phylogeographic data are necessary to: (1) to distinguish between gene pool subdivisions stemming from ancient phylogeographic splits and such originating from recent ecological divergence (Hewitt, 1996: Coyne and Orr, 2004, 2) to get to know a species' breeding system and dispersal biology and understand their likely roles in the origin and maintenance of reproductive isolation (Wendt et al., 2001, 2008) and (3) to assess the likely outcomes of population genetic processes in range margins (Bridle and Vines, 2007). We have carried out a range-wide population genetic and phylogeographic study of V. gigantea, a member of the Tillandsioideae subfamily of Bromeliaeceae, which has already attracted attention by evolutionary geneticists (Soltis et al., 1987; Barbará et al., 2007, 2008; Wendt et al., 2008).
Perhaps, the most outstanding features of our results are as follows. First, a decrease in genetic variability was observed from north to south, and this decrease was strongly correlated with latitude (Supplementary Figure 1). Second, the existence of a deep split between northern and southern populations of V. gigantea was shown by the cpDNA haplotypes (Figure 1), and this split coincides with the transition between clusters IV and V from allele frequencies at nuclear microsatellite loci (Figure 4). This discontinuity is located in the area occupied by today's Brazilian states of São Paulo and Rio de Janeiro at ca. 23 °S latitude (Figure 1). Third, cpDNA haplotypes revealed stronger genetic structure compared with the nuclear microsatellites (Table 5), and this difference allowed us to estimate the relative roles of pollen and seed dispersal in generating the observed genetic structure. Last, but not the least, decreased diversity and increased differentiation were observed in the range margins (Table 3), resulting in a marked increase in divergence (FST) and its variance in a range-wide isolation-by-distance analysis (Figure 2). The results are relevant to biogeography and conservation biology and to ongoing work on the evolutionary genetics of divergence in the adaptive radiation of Bromeliaceae.
Pleistocene population history
Chloroplast DNA variation in V. gigantea is geographically structured and the species is divided into two major phylogeographic groups, one in the north and one in the south (Figure 1). Despite obvious differences in the genetic patterns revealed by nuclear and cpDNA markers (Figures 1 and 4; Table 3), the two data sets contribute to a congruent picture of the history of this rainforest species and its constituent populations, as both highlight a historical division. The data would seem to support the hypothesis that V. gigantea survived the Pleistocene climatic oscillations in two fragmented refugia located between 24° and 20° latitude, comprising populations of JASP, BESP, PTRJ, RIRJ and SLES. One possible refugium was located slightly to the south of the phylogeographic split, close to populations BESP and JASP, from where the southward expansion would have taken place. For both nuclear and cpDNA, those two populations hold the greatest amount of genetic diversity (Table 3). The second putative refugium, as indicated by genetic diversity, was located in the northern region comprising populations of PTRJ, RIRJ and SLES. Note that populations of FLSC, PAPR and PTRJ are all located on islands a few kilometers off the coast, which explains their reduced cpDNA diversity despite high nuclear variability (see discussion of pollen/seed flow ratios below).
The two major haplogroups inferred from the cpDNA data have contrasting population histories in the Atlantic Rainforest. The shape of the haplotype network for the southern group (Figure 1b), where shorter branches are connected to the more widespread (and ancestral) haplotypes (H8 and H8), is indicative of recent population expansion. Greater haplotype divergence in the northern populations (Figure 1b) can be explained by the fact that these populations were likely submitted to cycles of long-term fragmentation and expansion due to climatic oscillations during the late Pleistocene. In contrast, the populations in the south were likely shaped by recent expansion and colonization of the vacant territory following the last glacial maximum. This scenario is in agreement with the latitudinal cline observed: a geographic trend of decreasing nuclear genetic diversity from north to south across the species' range (Table 3), indicated by a significant correlation between diversity and latitude (Supplementary Figure 1).
In the case of taxa with northern temperate distributions, rapid expansion from lower latitudes has been reported for many species, which explains the patterns of genetic divergence among extant populations (reviewed by Hewitt, 1996). In contrast, phylogeographic studies in southern and southeastern South America are rare and have thus far been limited to neotropical mammals (small mammals: Costa, 2003; Cerdocyon thous: Tchaicka et al., 2007), reptiles (Bothrops jararaca: Grazziotin et al., 2006) and birds (Xiphorhynchus fuscus: Cabanne et al., 2007, 2008). Few studies have reported phylogeography patterns in plants of the southern portion of the Brazilian Atlantic Rainforest, for example, those on Passiflora actinia and P. elegans from the Southern Brazilian States of Rio Grande do Sul, Santa Catarina and Paraná (Lorenz-Lemke et al., 2005) and for P. sellowii (Ledru et al., 2007). The phylogeographic patterns reported for these taxa are in agreement with those observed for V. gigantea. For B. jararaca, C. thous and P. elegans, recent demographic expansion was inferred for the southern groups, whereas northern populations seem to have enjoyed past demographic stability (Lorenz-Lemke et al., 2005; Grazziotin et al., 2006; Tchaicka et al., 2007). In our study the significant isolation by distance seen for nuclear microsatellites (Figure 2) and decreasing diversity from the north to south across the species' range (Supplementary Figure 1) are consistent with a biogeographic scenario in which the southern part of the Atlantic Rainforest was formed through migration of northern tropical elements from the Rio de Janeiro region (Rambo, 1951, 1960; Smith, 1962). The center of genetic diversity described here for V. gigantea—São Paulo and Rio de Janeiro (populations JASP, BESP, PTRJ and RIRJ; Figure 1)—coincides with the centers of species diversity and endemism for most animal and plant species from the Brazilian Atlantic Rainforest (Rambo, 1951; Smith, 1962; Moritz and Faith, 1998). This link between genetic and species diversity supports the hypothesis that both were shaped by the same biogeographic processes, triggered by the climatic oscillations of the Pleistocene. This hypothesis is based on the expectation that the dynamics of forest migration will be reflected by the demographic expansion of dependent organisms, such as the forest-dwelling V. gigantea.
The location of the north–south split in V. gigantea (Figure 1) does not coincide precisely with the latitudinal phylogeographic breaks reported by other phylogeographic studies of Atlantic Forest species (Costa, 2003; Lorenz-Lemke et al., 2005; Grazziotin et al., 2006; Cabanne et al., 2007, 2008; Tchaicka et al., 2007). This may be explained by the simple fact that barriers to dispersal will affect different species of animals and plants in different ways. Phylogeographic splits such as the ones found in this geographic region represent localities where cryptic species and incipient speciation may be detected in large-scale molecular systematic or ‘DNA barcoding’ studies (Lahaye et al., 2008). More phylogeographic work on species occurring in southern and southeastern Brazil is needed to shed light on the historical processes that have shaped the genetic diversity of the Atlantic Rainforest.
Patterns of diversity and gene flow and conclusions on pollen and seed dispersal
Our range-wide study in the neotropical rainforest species V. gigantea revealed remarkable differences in genetic structure among genomic compartments, with divergence (ΦST)=0.21 for nuclear DNA and (ΦST)=0.58 for cpDNA, translating into a pollen/seed flow ratio (Ennos, 1994) of ca. 3.3 to 1, and hence gene dispersal through pollen is threefold more efficient than dispersal through seeds in this species. The plumose seeds of V. gigantea are thought to be wind-dispersed and pollen grains are thought to be dispersed by bats (Sazima et al., 1999). These results agree well with data from a previous study for other species of the same subfamily of bromeliads (Tillandsioideae) based on trapping experiments (Cascante-Marín, 2005), and with recent estimates of pollen/seed flow ratios in the closely related genus Alcantarea based on fine-scale spatial genetic data (Barbará et al., 2008). The results indicate an important role for pollinators (that is, bats; Sazima et al., 1999) in maintaining population connectivity and species cohesion in V. gigantea, which has implications for both conservation and evolutionary biology. With respect to conservation, any disruption of the plant–pollinator mutualism will easily amplify inbreeding in this self-compatible species, and such disruptions have indeed been detected in the southern range margin (Paggi et al., 2007). With respect to evolutionary genetics, the important role of pollinators (that is, bats) in dispersal implies that any preference of pollinators for phenotypically divergent forms will easily contribute to disruption of gene flow and thus divergence, as discussed for bat-pollinated, color-polymorphic species of the closely related genus Alcantarea previously (Barbará et al., 2007, 2008). Similarly, a lack of preferences will lead to ‘leaky’ species barriers in the face of weak postyzygotic barriers in this group (Wendt et al., 2001, 2008; Barbará et al., 2007).
As was the case for the island populations, FLSC, PAPR and PTRJ, marginal populations of the current distribution range of V. gigantea (TMRS and SLES, Figure 1, Table 3) presented aberrant patterns of gene flow, diversity and differentiation when compared with central populations. Species margins are often seen as localities of central importance for population divergence and speciation (Mayr, 1954; Lesica and Allendorf, 1995; Bridle and Vines, 2007). The two peripheral populations (TMRS—southern margin, SLES—northern margin; Figure 1.) showed genetic features consistent with: (1) decreased genetic diversity (Table 3), (2) increased differentiation (Supplementary Table 2, Figure 2; but note that simple regression analysis may not be entirely adequate if there is autocorrelation in data and model residuals, see Diniz-Filho et al., 2009) and (3) limited gene flow compared with central populations (Figure 2; Figure 4).
Our data indicate the northern and southern range limits of V. gigantea as localities of interest to studies of divergence in range margins (Bridle and Vines, 2007). Of special relevance is that the two margins differ greatly in terms of their ecology: the northern margin coincides with a center of species diversity for this subfamily of bromeliads, whereas the southern margin is further south than the distribution limit of any other species in the genus. The hypothesis has been put forward that the evolutionary outcomes of reduced gene flow will differ between range margins with such contrasting ecological settings (Bridle and Vines, 2007). In-depth studies of this topic are only possible within a solid phylogeographic and population genetic framework such as the one provided here.
Implications for conservation
Our combined, range-wide chloroplast and nuclear DNA data allow us to suggest recommendations for ongoing conservation efforts in this world biodiversity ‘hotspot’. In situ and ex situ conservation efforts should explicitly consider the two biogeographic units indicated by our data. The most effective conservation strategy for V. gigantea may consist of sampling populations from each of the six clusters identified by the Bayesian genetic structure analysis. Priority should be given to populations of JASP and BESP, which showed the highest values of nuclear and cpDNA diversity. Chloroplast DNA-based assessment of genetic variability for coastal island populations may be misleading because of restricted seed dispersal as indicated by population of PTRJ, which highlights the need to consider nuclear and cpDNA data simultaneously. The relatively clear gene pool structure observed may allow the tracking of plant material used in horticulture by using genetic assignment tests to detect illegal harvesting of plants from nature.
This study shows how the comparative analysis of maternally inherited cpDNA markers and biparentally inherited nuclear markers can provide a deeper understanding of the dynamics responsible for both ancient and more recent events that have shaped the current distribution of genetic variability in neotropical plants. Combined data sets of this type are available for many northern temperate taxa, but information for neotropical species is extremely scarce. The results are relevant to conservation efforts and ongoing work on the genetics of population divergence and speciation in this biodiversity-rich region of the neotropics.
References
Aleixo A (2004). Historical diversification of a Terra-firme forest bird superspecies: a phylogeographic perspective on the role of different hypotheses of Amazonian diversification. Evolution 58: 1303–1317.
Bandelt HJ, Forster P, Rohl A (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48.
Barbará T, Lexer C, Martinelli G, Mayo S, Fay MF, Heuertz M (2008). Within-population spatial genetic structure in four naturally fragmented species of a neotropical inselberg radiation, Alcantarea imperialis, A. geniculata, A. glaziouana and A. regina (Bromeliaceae). Heredity 101: 285–296.
Barbará T, Martinelli G, Fay MF, Mayo SJ, Lexer C (2007). Population differentiation and species cohesion in two closely related plants adapted to neotropical high-altitude ‘inselbergs’, Alcantarea imperialis and Alcantarea geniculata (Bromeliaceae). Mol Ecol 16: 1981–1992.
Benzing D (2000). Bromeliaceae: Profile of an Adaptive Radiation. Cambridge University Press: Cambridge, UK.
Boneh L, Kuperus P, Van Tienderen PH (2003). Microsatellites in the bromeliads Tillandsia fasciculata and Guzmania monostachya. Mol Ecol Notes 3: 302–303.
Bridle JR, Vines TH (2007). Limits to evolution at range margins: when and why does adaptation fail? Trends Ecol Evol 22: 140–147.
Butlin RK, Ritchie MG (2009). Genetics of speciation. Heredity 102: 1–3.
Cabanne GS, d'Horta FM, Sari EHR, Santos FR, Miyaki CY (2008). Nuclear and mitochondrial phylogeography of the Atlantic forest endemic Xiphorhynchus fuscus (Aves: Dendrocolaptidae): biogeography and systematics implications. Mol Phylogenet Evol 49: 760–773.
Cabanne GS, Santos FR, Miyaki CY (2007). Phylogeography of Xiphorhynchus fuscus (Passeriformes, Dendrocolaptidae): vicariance and recent demographic expansion in southern Atlantic forest. Biol J Linn Soc 91: 73–84.
Carnaval AC, Moritz C (2008). Historical climate modelling predicts patterns of current biodiversity in the Brazilian Atlantic forest. J Biogeogr 35: 1187–1201.
Cascante-Marín A (2005). Establishment, reproduction and genetics of epiphytic bromeliad communities during premontane forest succession in Costa Rica. Doctoral Thesis, University of Amsterdam, The Netherlands.
Cavallari MM, Forzza RC, Veasey EA, Zucchi MI, Oliveira GCX (2006). Genetic variation in three endangered species of Encholirium (Bromeliaceae) from Cadeia do Espinhaco, Brazil, detected using RAPD markers. Biodivers Conserv 15: 4357–4373.
Costa LP (2003). The historical bridge between the Amazon and the Atlantic forest of Brazil: a study of molecular phylogeography with small mammals. J Biogeogr 30: 71–86.
Coyne JA, Orr HA (2004). Speciation. Sinauer Associates: Sounderland, MA.
da Silva JMC, de Sousa MC, Castelletti CHM (2004). Areas of endemism for passerine birds in the Atlantic forest, South America. Glob Ecol Biogeogr 13: 85–92.
Dieringer D, Schlotterer C (2003). Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 3: 167–169.
Diniz-Filho JAF, Nabout JC, Telles MPC, Soares TN, Rangel TFLVB (2009). A review of techniques for spatial modeling in geographical, conservation and landscape genetics. Genet Mol Biol 32: 203–211.
El Mousadik A, Petit RJ (1996). High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L) Skeels] endemic to Morocco. Theor Appl Genet 92: 832–839.
Ennos RA (1994). Estimating the relative rates of pollen and seed migration among plant-populations. Heredity 72: 250–259.
Ennos RA, Sinclair WT, Hu X-S, Langdon A (1999). Using organelle markers to elucidade the history, ecology, and evolution of plant populations. In: Hollingsworth PM, Bateman RM, Gornall RJ (eds). Molecular Systematics and Plant Evolution. Taylor & Francis: London, pp 1–19.
Evanno G, Regnaut S, Goudet J (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Laval LG, Schneider S (2005). Arlequin version 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47–50.
Goudet J (1995). FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86: 485–486.
Grazziotin FG, Monzel M, Echeverrigaray S, Bonatto SL (2006). Phylogeography of the Bothrops jararaca complex (Serpentes : Viperidae): past fragmentation and island colonization in the Brazilian Atlantic Forest. Mol Ecol 15: 3969–3982.
Hewitt GM (1996). Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58: 247–276.
Hewitt GM (2004). A climate for colonization. Heredity 92: 1–2.
Hwang LH, Hwaing SY, Lin TP (2000). Low chloroplast DNA variation and population differentiation of Chamaecypaeis formosensis and Chamaecypaeis taiwanensis. Taiwan Lin Ye Ke Xue 15: 229–236.
Lahaye R, Van der Bank M, Bogarin D, Warner J, Pupulin F, Gigot G et al. (2008). DNA barcoding the floras of biodiversity hotspots. Proc Natl Acad Sci USA 105: 2923–2928.
Ledru MP, Salatino MLF, Ceccantini G, Salatino A, Pinheiro F, Pintaud JC (2007). Regional assessment of the impact of climatic change on the distribution of a tropical conifer in the lowlands of South America. Divers Distrib 13: 761–771.
Lesica P, Allendorf FW (1995). When are peripheral-populations valuable for conservation. Conserv Biol 9: 753–760.
Lorenz-Lemke AP, Muschner VC, Bonatto SL, Cervi AC, Salzano FM, Freitas LB (2005). Phylogeographic inferences concerning evolution of Brazilian Passiflora actinia and P. elegans (Passifloraceae) based on ITS (nrDNA) variation. Ann Bot 95: 799–806.
Mayr E (1954). Change of genetic environment and evolution. In: Huxley JS, Hardy AC, Ford EB (eds). Evolution as a Process. Allen & Unwin: London, pp 156–180.
Moritz C, Faith DP (1998). Comparative phylogeography and the identification of genetically divergent areas for conservation. Mol Ecol 7: 419–429.
Myers N, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Kent J (2000). Biodiversity hotspots for conservation priorities. Nature 403: 853–858.
Nei M (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590.
Paggi GM, Palma-Silva C, Silveira LCT, Kaltchuk-Santos E, Bodanese-Zanettini MH, Bered F (2007). Fertility of Vriesea gigantea Gaud. (Bromeliaceae) in Southern Brazil. Am J Bot 94: 683–689.
Palma-Silva C, Cavallari MM, Barbará T, Lexer C, Gimenes MA, Bered F et al. (2007). A set of polymorphic microsatellite loci for Vriesea gigantea and Alcantarea imperialis (Bromeliaceae) and cross-amplification in other bromeliad species. Mol Ecol Notes 7: 654–657.
Palma-Silva C, Paggi GM, Felicetti RA, Ferraz R, Kaltchuk-Santos E, Bered F et al. (2008). Meiotic behavior and pollen viability of wild populations of the neotropical species Vriesea gigantea (Bromeliaceae). Plant Species Biol 23: 217–221.
Pennington RT, Lavin M, Prado DE, Pendry CA, Pell SK, Butterworth CA (2002). Annual Meeting of the Association-for-Tropical-Biology-and –Conservation, Panama City, Panama. pp 515–537.
Petit RJ, El Mousadik A, Pons O (1998). Identifying populations for conservation on the basis of genetic markers. Conserv Biol 12: 844–855.
Pritchard JK, Stephens M, Donnelly P (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Rambo B (1951). A imigração da selva higrófila no Rio Grande do Sul. Sellowia 3: 55–91.
Rambo B (1960). Die Sudgrenze des brasilianschen regenwaldes. Pesquisas 8: 5–41.
Raymond M, Rousset F (1995). GENEPOP (version-1.2)—population-genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Rull V (2006). Quaternary speciation in the neotropics. Mol Ecol 15: 4257–4259.
Rull V (2008). Speciation timing and neotropical biodiversity: the Tertiary-Quaternary debate in the light of molecular phylogenetic evidence. Mol Ecol 17: 2722–2729.
Sang T, Crawford DJ, Stuessy TF (1997). Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am J Bot 84: 1120–1136.
Sarthou C, Samadi S, Boisselier-Dubayle MC (2001). Genetic structure of the saxicole Pitcairnia geyskesii (Bromeliaceae) on inselbergs in French Guiana. Am J Bot 88: 861–868.
Sazima M, Buzato S, Sazima I (1999). Bat-pollinated flower assemblages and bat visitors at two Atlantic forest sites in Brazil. Ann Bot 83: 705–712.
Smith LB (1962). Origins of the flora of southern Brazil. Contr U.S. Natl Herb 35: 215–227.
Smith LB, Downs RJ (1977). Bromeliaceae (Tillandsioideae) Flora Neotropica Monografia. Hafner Press: New York, USA.
Sokal RR, Rohlf FI (1995). Biometry: The Principles and Practice of Statistics in Biological Research. Freeman and Company: New York.
Soltis DE, Gilmartin AJ, Rieseberg L, Gardner S (1987). Genetic variation in the epiphytes Tillandsia ionantha and Tillandsia recurvata (Bromeliaceae). Am J Bot 74: 531–537.
Speranza PR, Seijo JG, Grela IA, Neffa VGS (2007). Chloroplast DNA variation in the Turnera sidoides L. complex (Turneraceae): biogeographical implications. J Biogeogr 34: 427–436.
Taberlet P, Gielly L, Pautou G, Bouvet J (1991). Universal primers for amplification of three noncoding regions of chloroplast DNA. Plant Mol Biol 17: 1105–1109.
Tchaicka L, Eizirik E, De Oliveira TG, Candido JF, Freitas TRO (2007). Phylogeography and population history of the crab-eating fox (Cerdocyon thous). Mol Ecol 16: 819–838.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Wendt T, Canela MBF, De Faria APG, Rios RI (2001). Reproductive biology and natural hybridization between two endemic species of Pitcairnia (Bromeliaceae). Am J Bot 88: 1760–1767.
Wendt T, Coser TS, Matallana G, Guilherme FAG (2008). An apparent lack of prezygotic reproductive isolation among 42 sympatric species of Bromeliaceae in southeastern Brazil. Plant Syst Evol 275: 31–41.
Widmer A, Lexer C, Cozzolino S (2009). Evolution of reproductive isolation in plants. Heredity 102: 31–38.
Wright S (1965). The interpretation of population structure by F-statistics with special regards to system of mating. Evolution 19: 395–420.
Zaú AS (1998). Fragmentação da Mata Atlântica: aspectos teóricos. Floresta e Ambiente 5: 160–170.
Acknowledgements
We thank Silvia Richter, Jamilla Sampaio, Jeffrey Joseph, Laura Kelly and Rhian Smith for their help in the laboratory, Simon Mayo and Jon Bridle for their helpful comments on earlier versions of this paper and IBAMA for processing of collection/export permits. We are grateful to all colleagues who helped us during the field work. This work was supported by CNPq, CAPES and by awards from the Andrew W Mellon Foundation to CPS and GMP and by Weston Foundation to TB.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Heredity website (http://www.nature.com/hdy)
Supplementary information
Rights and permissions
About this article
Cite this article
Palma-Silva, C., Lexer, C., Paggi, G. et al. Range-wide patterns of nuclear and chloroplast DNA diversity in Vriesea gigantea (Bromeliaceae), a neotropical forest species. Heredity 103, 503–512 (2009). https://doi.org/10.1038/hdy.2009.116
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2009.116
Keywords
This article is cited by
-
Genetic diversity and population structure of Aechmea distichantha (Bromeliaceae), a widely geographically distributed species in South America
Plant Systematics and Evolution (2023)
-
Patterns of genetic diversity and structure of a threatened palm species (Euterpe edulis Arecaceae) from the Brazilian Atlantic Forest
Heredity (2022)
-
From micro- to macroevolution: insights from a Neotropical bromeliad with high population genetic structure adapted to rock outcrops
Heredity (2020)
-
Genetic diversity and karyotype of Pitcairnia azouryi: an endangered species of Bromeliaceae endemic to Atlantic Forest inselbergs
Molecular Biology Reports (2020)
-
Genetic structure and gene flow among populations of Encholirium magalhaesii, a rocky grassland fields bromeliad
Brazilian Journal of Botany (2020)
