
Abstract
Injecting proteins into the central nervous system that stimulate neuronal growth can lead to beneficial effects in animal models of disease. In particular, glial cell line-derived neurotrophic factor (GDNF) has shown promise in animal and cell models of Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis (ALS). Here, systemic AAV9-GDNF was delivered via tail vein injections to young rats to determine whether this could be a safe and functional strategy to treat the SOD1G93A rat model of ALS and, therefore, translated to a therapy for ALS patients. We found that GDNF administration in this manner resulted in modest functional improvement, whereby grip strength was maintained for longer and the onset of forelimb paralysis was delayed compared to non-treated rats. This did not, however, translate into an extension in survival. In addition, ALS rats receiving GDNF exhibited slower weight gain, reduced activity levels and decreased working memory. Collectively, these results confirm that caution should be applied when applying growth factors such as GDNF systemically to multiple tissues.
Similar content being viewed by others

Introduction
Amyotrophic lateral sclerosis (ALS) is characterized by progressive loss of upper and lower motor neurons, typically leading to muscle atrophy, paralysis and death within 3–5 years of diagnosis.1 Most ALS cases are of unknown etiology and sporadic in nature (90–95%) with no genetic association. However, familial ALS also exists and is associated with genes such as Cu/Zn superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TARDP), C9ORF72(2, 3) and NEK1.4, 5 After a half century of trials and testing of over 150 therapeutic agents or strategies in pre-clinical models, there are still no successful treatment options for ALS, with the single developed drug, Riluzole, prolonging survival by only ~2–3 months.
Drug development for ALS has been limited because most drugs do not cross the Basso, Beattie and Breshnahan (BBB) and hence cannot enter the central nervous system (CNS). Fortunately, pre-clinical animal studies have shown that viral vectors can cross the BBB in order to deliver drugs to the CNS, providing promising options for gene therapy. Adeno-associated virus (AAV) vectors have become a popular vehicle of choice for drug and growth factor delivery for the treatment of neurological disorders.6 AAV serotype 2 vectors are in clinical trials for Parkinson’s disease, with patients receiving direct cranial delivery without any serious adverse events attributable to the vector.7, 8, 9, 10 Indeed, a completed AAV serotype 2–glutamic acid decarboxylase gene therapy trial for Parkinson’s disease was the first phase 2 double-blinded clinical study to demonstrate the efficacy of gene therapy in a neurological disorder.9
AAV serotype 9 (AAV9) is an additional promising viral vector for gene delivery, as it crosses the BBB, has a high gene transfer efficiency in the brain11 and transduces motor neurons and astrocytes.12, 13 Using AAV9-SOD1-shRNA to knock down mutant SOD1 significantly delayed disease onset and extended survival both in SOD1 mice receiving peripheral injections and in SOD1 rats receiving direct targeting of the motor cortex.14, 15 Importantly, intrathecal delivery of this AAV9-SOD1-shRNA to non-human primates was proven safe and effective in suppressing SOD1 expression in motor neurons and glia throughout the spinal cord.14 Furthermore, AAV9 delivery of microRNA via intracerebral ventricular injection in neonatal SOD1 mice resulted in a 50% extension in survival.16 Rescue of the spinal muscular atrophy phenotype in mice was also demonstrated following AAV9 delivery of the SMN gene.17 This strategy for delivering SMN has been deemed safe in larger animal models18 and non-human primates,19 and is now being used in clinical trials for spinal muscular atrophy patients. Together, these studies set the stage for AAV9-mediated therapy in human clinical trials.
Glial cell line-derived neurotrophic factor (GDNF) is a powerful growth factor that can protect both dopamine and motor neurons in vitro.20, 21, 22 However, delivery to the CNS has been challenging given that it does not cross the BBB. Using a variety of delivery methods, including direct protein, stem cell and viral, GDNF has been shown to protect dopamine and motor neurons in a number of animal models.23, 24, 25, 26 GDNF delivered into the spinal cord protected spinal motor neurons in ALS rats; however, it did not preserve neuromuscular junctions or prolong lifespan, perhaps as delivery was limited to the spinal cord.27 In contrast, GDNF treatment in the muscle of ALS transgenic mice and rats was able to preserve neuromuscular junctions and protect motor neurons, presumably due to retrograde transport.27, 28, 29, 30 Importantly, AAV-GDNF delivery to the muscle of ALS mice provided not only neuroprotection but also functional effects.31, 32, 33, 34
The goal of the current study was to determine if global expression of GDNF in multiple tissues could have a beneficial effect in the SOD1 rat model of ALS. To achieve this we used the capacity of AAV9 to cross the BBB in order to deliver GDNF to young SOD1G93A (herein, SOD1) ALS rats using tail vein injection, which in other studies has led to widespread expression of the vector.12, 14, 17 GDNF administration in this manner resulted in delivery to many tissues including brain, spinal cord and muscle. However, this led to only modest functional improvements in SOD1 rats, did not translate into an extension in lifespan and was associated with several side effects including slow weight gain, reduced activity and working memory deficits. It is therefore important to consider that although GDNF can support some populations of neurons to provide a beneficial effect for neurodegenerative diseases, administration should be targeted to specific cellular populations in order to avoid widespread side effects.
Results
GDNF is expressed in the brain, spinal cord and muscle following systemic AAV9 tail vein delivery
SOD1 and wild-type (WT) rats received tail vein injections of AAV9-GDNF or AAV9 alone [‘AAV9(−)’] at postnatal (p) day 25. Histological analysis of brain and cervical spinal cord tissue at disease end point of AAV9-GDNF-treated SOD1 animals revealed GDNF staining in the cortex, hippocampus and septal region (Figure 1a). There was also GDNF staining in the spinal cord gray matter, which showed a similar distribution in rats euthanized at a short time point (4-week post injection). GDNF was not observed in the AAV9(−)-treated SOD1 animals brain or spinal cord tissue. A sensitive ELISA assay confirmed that WT and SOD1 rats administered AAV9-GDNF showed significantly more GDNF expression in the brain, cervical and lumbar spinal cord as well as hindlimb and forelimb muscles but not in the kidney, relative to AAV9(−) (Figure 1b). In contrast, GDNF was undetectable in the serum of any group at study end point.

Increased GDNF expression in CNS tissue following systemic injection of AAV9-GDNF. (a) Histological analysis of GDNF expression in CNS tissue sections revealed GDNF staining in the cortex, septum and hippocampus, as well as the spinal cord gray matter in rats injected with AAV9-GDNF but not those injected with AAV9(−) control virus. (b) Quantification of GDNF expression in various tissues from both WT and SOD1 rats by ELISA confirmed that after injections of AAV9-GDNF, GDNF levels were increased in the brain, spinal cord and muscle relative to AAV9(−)-injected controls but not in the kidney. There were no detectable levels of GDNF expression in the serum of any of the groups. (c) Immunostaining for ChAT in the ventral horn of the cervical spinal cord revealed significantly greater numbers of (d) total ChAT+ cells and (e) ChAT+ cells >700 μm per section in AAV9-GDNF-treated SOD1 rats relative to AAV9(−) controls. (f) Average ChAT+ cell size was not significantly different. Sample size: n=4 per group and the same animals were used for each assay. *P<0.05, unpaired, two-tailed t-test; error bars=s.e.m. Scale bars a, b: 500 μm; c: 100 μm. ELISA, enzyme-linked immunosorbent assay; FL, forelimb; HL, hindlimb; SC, spinal cord.
Consistent with the previous reports that GDNF protects spinal motor neurons,27 analysis of ventral horn choline acetyl transferase+ (ChAT+) cells in the cervical spinal cord revealed a significant increase in total numbers of ChAT+ cells and total large (>700 μm) ChAT+ cells, but not average ChAT+ cell size in the ventral horn of AAV9-GDNF-treated SOD1 rats relative to AAV9(−)-treated SOD1 rats euthanized at end point (Figures 1c–f).
GDNF maintains forelimb strength and delays onset of forelimb paralysis in SOD1 rats, but does not effect survival
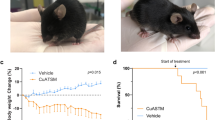
SOD1 rats underwent grip strength testing in order to assess forelimb and hindlimb strength. SOD1 rats that were administered AAV9-GDNF at p25 showed a significant increase in forelimb, but not hindlimb, grip strength relative to AAV9(−) SOD1 rats over time (Figures 2a and b, respectively). AAV9-GDNF administration in WT rats did not appear to improve grip strength in either forelimb or hindlimb regions relative to saline and AAV9(−) controls (Figures 2c and d, respectively). SOD1 rats also underwent locomotor scoring in order to assess forelimb and hindlimb motor function deficits (Figures 2e and f, respectively). Rats that were administered AAV9-GDNF showed a significant delay in forelimb onset relative to AAV9(−) controls (Figures 2e and g); however, hindlimb function was not affected (Figure 2f). Both AAV9-GDNF and AAV9(−) controls showed similar survival times, demonstrating that the delay in disease onset did not translate to an extension in survival (Figure 2h).

GDNF administration leads to modest functional improvements in SOD1 rats. SOD1 rats administered AAV9-GDNF showed enhanced (a) forelimb but not (b) hindlimb grip strength over time relative to SOD1 AAV9(−)-injected controls (*P<0.05, two-way analysis of variance (ANOVA) error bars=s.e.m.). GDNF administration in WT rats did not lead to improved (c) forelimb or (d) hindlimb grip strength relative to saline- or (AAV9(−)-injected controls. (e) Forelimb and (f) hindlimb BBB scoring was performed to assess motor function deficits in SOD1 rats, and while there were no differences in hindlimb motor performance, (e, g) forelimb onset was significantly delayed after AAV9-GDNF administration relative to AAV9(−) controls (*P<0.05, Wilcoxon signed rank test). Differences in time of onset did not however translate into extended survival (h) as there was no difference between survival times in AAV9-GDNF- and AAV9(−)-injected SOD1 rats. Given that 70% of AAV9(−) and 60% of AAV9-GDNF rats showed onset in both limbs at the same time point or within 7 days of one another, and that each rat at end point exhibited a significant degree of both forelimb and hindlimb paralysis, rats were not further separated into groups based on fore- or hindlimb onset *P<0.05, two-way ANOVA error bars=s.e.m.
GDNF lowers body weight, reduces open field activity and leads to reduction in working memory in both WT and SOD1 rats
Monitoring weight gain in WT rats that received AAV9-GDNF, AAV9(−) or saline at p25 showed that all rats gained weight over time; however, rats receiving AAV9-GDNF gained weight at a significantly slower rate relative to rats receiving saline or AAV9(−) control virus (Figure 3a). Both SOD1 and WT rats receiving AAV9-GDNF showed a significantly slower weight gain relative to SOD1 AAV9(−) controls (Figure 3b). In contrast to WT AAV9-GDNF rats that continued to gain weight, SOD1 rats receiving AAV9(−) or AAV9-GDNF reached their peak body weight and then steadily lost weight as signs of paralysis and disease progression were observed. Interestingly, as age of peak body weight is often used as an indicator of age of disease onset, although GDNF slowed the weight gain in both WT and SOD1 rats relative to AAV9(−) controls, we found that SOD1 rats that were administered AAV9-GDNF reached their peak body weight at 162.9±7.6 days, which was a significantly later time than SOD1 rats injected with AAV9(−) control virus at 140.7±4.7 (Figure 3b), suggestive of a delayed disease onset.

GDNF administration leads to slower weight gain, reduction in activity and deficits in working memory. (a) WT and (b) SOD1 rats that received tail vein injections of AAV9-GDNF gained weight at a slower rate over time relative to their AAV9(−)- and saline- injected controls. (WT saline vs WT AAV9-GDNF *P<0.05, two-way analysis of variance (ANOVA): interaction, time, group; WT AAV9(−) vs AAV9-GDNF *P<0.05, two-way ANOVA: interaction, time. SOD1 AAV9(−) and SOD1 AAV9-GDNF vs WT AAV9-GDNF *P<0.05, two-way ANOVA: interaction, time, group; SOD1 AAV9(−) vs SOD1 AAV9-GDNF *P<0.05, two-way ANOVA: interaction, time). (c) When placed in an open field apparatus for 30 min, both WT and SOD1 rats that received AAV9-GDNF had a significantly reduced total activity level relative to their respective AAV9(−) controls. In addition, SOD1 AAV9(−)-injected rats showed significantly lower total activity relative to WT AAV9(−)-injected rats that would suggest that these SOD1 rats are in the beginning stages of disease. When tested for working memory in the Y-Maze task, rats injected with AAV9-GDNF exhibited significant working memory deficits (d) as the % alternation between arms was significantly reduced in both WT and SOD1 rats relative to AAV9(−) controls. (e) This was not due to a decrease in total observed activity in the Y-Maze, as overall arm entries between AAV9-GDNF and AAV9(−) groups were not different. *P<0.05, **P<0.01, ***P<0.0001, error bars=s.e.m., unpaired, two-tailed t-test.
Following systemic delivery of either AAV9-GDNF or AAV9(−) at p25, both WT and SOD1 rats at p125 receiving GDNF showed signs of significantly reduced overall activity when placed in an open field testing apparatus, relative to their respective AAV9(−) controls (Figure 3c). Relative to WT AAV9(−) controls, SOD1 AAV9(−) controls showed a significantly reduced activity level, which is likely indicative of the initial stage of ALS disease symptoms. When assessed in the Y-Maze, rats receiving GDNF alternated between arms significantly less than rats receiving AAV9(−) control virus, suggesting a reduction in working memory (Figure 3d). There were no differences in working memory between WT and SOD1 groups. The total entries into each arm did not differ among the groups (Figure 3e), demonstrating that the reduced alternation between rats receiving GDNF and rats receiving AAV9(−) was not due to differences in the overall ability of the rats to perform the task.
Discussion
Therapeutic delivery using AAV9 has been applied in the treatment of various neurodegenerative diseases including ALS, spinal muscular atrophy, PD35 and Huntington’s disease.36 This strategy has been deemed safe in rodents,17 pigs18 and non-human primates,19 and is now being used in clinical trials for spinal muscular atrophy patients. GDNF delivery to neurons of the CNS has been difficult as the protein does not cross the BBB. To circumvent this issue, different modes of delivery (cannula, viral or modified cells) into various CNS regions (intraventricular, intrastriatal or intranigral) have been studied. Here, we assessed systemic delivery of GDNF using AAV9 tail vein injections in the SOD1 rat model of ALS.
In the current study, we show that systemic injection of AAV9-GDNF confers modest functional improvements in the SOD1 ALS rat, which included increased forelimb strength and delayed onset of forelimb paralysis. We have previously shown that GDNF delivery to the spinal cord can protect motor neurons.27, 30 The current study, however, did not assess motor neurons and neuromuscular junctions as animal tissue was collected at end point, when motor neurons and neuromuscular junctions are fully degenerated.15 Although systemically delivered AAV9-GDNF provided some functional effects, there were concomitant side effects in both WT and SOD1 rats and no extension in lifespan. Side effects included slower weight gain, reduction in open field activity and reduction in working memory. Weight loss has been reported as a minor side effect in a number of studies following AAV serotype 2–GDNF delivery to rodents and non-human primates.37, 38, 39 When clinical trials for Parkinson’s disease infused GDNF protein into the ventricles, patients also demonstrated side effects of weight loss, along with Lhermittes, a condition described as an uncomfortable electrical sensation that runs down the back and into the limbs.40, 41 Weight loss, at least in rats, was attributed to high GDNF protein levels in the substantia nigra with subsequent transport and secretion of GDNF in the hypothalamus, activating a population of corticotrophin-releasing factor neurons.37 In a different open label trial performed by our group and subsequently others, GDNF was infused directly into the caudate putamen.42, 43 These clinical studies showed excellent tolerance, a clinical benefit and few side effects that predominantly included Lhermittes. Unfortunately, a follow-up double-blind clinical trial showed that GDNF delivered to the caudate putamen did not confer the predetermined level of benefit for Parkinson’s disease patients; however, this result was likely due to the mode of delivery.44 Critically, the collective clinical trials all confirmed that local GDNF delivery was safe for patients with no overt toxic effects.
Results from the current and previous studies highlight that the appropriate delivery method, tissue targeting and targeting of specific cell types within the tissue are critical for maximizing the potential for GDNF-mediated therapeutic benefit. For instance, GDNF delivery to the muscle of ALS mice had profound neuroprotective effects on both neuromuscular junctions and spinal motor neurons.31, 33 Furthermore, transgenic GDNF expression from birth in the muscle fibers of ALS mice resulted in delayed disease onset and prolonged survival, whereas transgenic GDNF expression in astrocytes did not affect disease.45 On the other hand, GDNF delivery by direct intraspinal injection of lentivirus encoding GDNF into the SOD1 mouse did not prevent the loss of spinal motor neurons.46 In addition, transplanting non-engineered human neural progenitor cells that differentiate into astrocytes in the spinal cord of SOD1 rats was not neuroprotective.27 In contrast, when human neural progenitor cells were genetically engineered to overexpress GDNF and then transplanted into the spinal cord to become astrocytes, they were able to protect motor neurons in the SOD1 rat and also following nerve axotomy.30, 47 This critically supports the idea that there is a synergistic effect when providing both GDNF and healthy progenitor cells/astrocytes. In the current study, consistent with reported location of known GDNF receptors,48, 49 GDNF protein levels were significantly elevated in the brain, spinal cord and muscle. Although the systemic delivery of GDNF helped to enhance cervical spinal motor neuron survival, maintain forelimb grip strength and delay the onset of forelimb paralysis in SOD1 rats, it did not effect survival. Data would suggest that the protection of spinal motor neurons is not enough in itself to prevent disease onset or halt progression. As the mechanisms underlying the origin and progression of ALS remain elusive, it is difficult to speculate which cells in the CNS might benefit the most from GDNF exposure and at what time point during disease. It may be that greater GDNF expression levels in specific cell types within various CNS regions are needed, or particular delivery methods and targeting strategies are necessary for a more pronounced therapeutic benefit.
We have demonstrated that systemic GDNF was associated with certain side effects including slowed weight gain, reduced overall activity levels and impaired working memory. However, these side effects may be acceptable if GDNF could significantly extend lifespan of ALS patients. Unfortunately, we show that systemic injection of AAV9-GDNF confers only modest functional improvements in the SOD1 ALS rat and no extension in lifespan. Collectively, these results lead us to conclude that AAV9-based systemic delivery of GDNF would not likely be a useful strategy for targeting ALS in the clinic.
Rather than systemic GDNF delivery, patients may benefit from targeted delivery to specific regions of the CNS using direct viral infection or a pump. However, lentiviral vectors to deliver GDNF to spinal motor neurons have not been sufficient to prevent motor neuron loss or muscle denervation in ALS mice.46 In contrast, a combined ex vivo approach involving the delivery of GDNF with genetically engineered human neural progenitor cells that generate astrocytes following grafting was shown to protect dying spinal motor neurons in ALS rats27, 30 and also dopamine neurons in Parkinson’s rats.50 The data suggests that targeted growth factor delivery combined with new support cells serving as minipumps may be the most powerful gene and cellular therapeutic strategy for various neurodegenerative diseases. This approach is now being pursued in the first ever cell and gene therapy trial for ALS (ClinicalTrials.gov Identifier: NCT02943850).
Materials and methods
Animals
WT and SOD1G93A (‘SOD1’) transgenic rats (Sprague–Dawley background) were housed under NIH guidelines and all experiments were conducted in accordance with the Cedars-Sinai Institutional Animal Care and Use Committee (IACUC protocol 4260), and the Guide for the Care and Use of Laboratory Animals. This colony provides later onset and end point than the original published model.51 Reminiscent of human pathology, disease onset in hindlimbs and/or forelimbs is unpredictable in this model with overt paresis progressing to complete paralysis. Male and female rats showed no significant differences in either WT or SOD1 rats; therefore data were pooled for each genotype.
Injections
At ~p25, rats were administered tail vein injections of either empty AAV9 virus [‘AAV9(−)’] (SOD1, n=10) or AAV9-GDNF (SOD1, n=8; WT, n=4). GDNF expression was driven by the cytomegalovirus enhancer/chicken beta-actin promoter. In a second experiment, WT rats only were administered tail vein injections of either saline (n=6), AAV9(−) (n=6) or AAV9-GDNF (n=6). Rats were injected with 100 μl of viral solution containing a mixture of phosphate buffered saline and 4 × 1012 DNase-resistant particles.
Behavioral assessment
At ~70 days, rats underwent weekly body weight assessment. Behavioral testing started at ~110 days (presymptomatic for SOD1) and included forelimb/hindlimb grip strength assessment and forelimb/hindlimb BBB locomotor rating scale scoring.52 Open field tests for 30 min and Y-Maze tests for 10 min were conducted once, when rats were ~125 days of age. SOD1 rats were followed to disease end point to determine whether global GDNF ameliorates disease progression. WT rats were examined to ~200 days to determine whether systemic GDNF had any adverse effects in non-diseased rats.
Assessment of motor behavior
The BBB locomotor rating scale52 assesses an animal’s ability to walk and has been used to quantify limb paralysis. The 21-point BBB scoring is an open field locomotor test of limb function that provides an indication of when paralysis starts in any limb and the degree of progression continuing until the animal's end point. At ~70 days and continuing until disease end point, an observer blinded for genotype and treatment assessed animals once or twice weekly for BBB scores and body weights in order to determine the average age of disease onset and survival. If left and right limb scores were different, the average for each (hind/fore) limb was taken as the rat’s overall score. Disease onset was when an animal displayed a BBB score of 15 or lower. End point was classified as severe forelimb and/or hindlimb paralysis resulting in the animal’s inability to right itself within 25 s after being placed on either one of its sides. Given that 70% of AAV9(−) and 60% of AAV9-GDNF rats showed onset in both limbs at the same time point or within 7 days of one another, and that each rat at end point exhibited a significant degree of both forelimb and hindlimb paralysis, rats were not further separated into groups based on fore- or hindlimb onset. Male and female rats did not differ significantly in BBB scoring, grip strength, open field or Y-Maze and data were therefore pooled. To normalize for gender differences in body weight, baseline weight measurements were taken at 70 days and the percent of body weight gain from day 70 was used for data analysis. Male and female percent body weight gain was not significantly different and the data were pooled.
Tissue analysis
Animals were euthanized at study end point with ketamine/xylazine and transcardially perfused with 0.9% saline. A small cohort of rats was euthanized at 4 weeks post injection in order to assess early AAV9 and GDNF distribution. The brain, spinal cord and muscle tissue were collected by cutting down the midline of each sample so that both histological and protein analysis could be performed. Samples collected for protein analysis were fast-frozen in liquid nitrogen and stored at −80 °C. Samples collected for histological analysis were placed in 4% paraformaldehyde, post-fixed overnight and transferred to 30% sucrose. For immunohistochemistry, brain and spinal cords were sectioned at 35 μm using a microtome and collected as free-floating sections, and muscle tissue was sectioned, using a cryostat, directly onto pre-coated slides at 25 μm. Tissue was stained using anti-GDNF (1:100, purified goat IgG, Cat # AF-212-NA, R&D Systems, Minneapolis, MN, USA).
Analysis of spinal motor neurons
Spinal cord sections were immunostained for spinal motor neurons using an antibody against ChAT (goat, 1:250, Millipore, Billerica, MA, USA), and counterstained with 4',6-diamidino-2-phenylindole. One 20 × image stack per section (six sections total, 420 μm apart) was captured encompassing the lateral ventral horn of the cervical (C4-C7) spinal cord. ChAT+ cells were counted in order to determine total spinal motor neuron numbers, and cell body size was measured using Image J software.
ELISA GDNF protein assay
Homogenized tissues at 100 mg ml-1 in 1 × phosphate buffered saline+0.5% Triton X-100+protease inhibitor cocktail (Roche, Indianapolis, IN, USA) were centrifuged at 4 °C at 4000 rpm for 5 min. Supernatant was collected and stored at −80 °C. DuoSet ELISA kit determined GDNF concentration (DY212, R&D systems) according to the manufacturer’s instructions. Samples were run with biological (n>4 per group) and technical (n=3) replicates.
Statistical analysis
Statistical analyses were performed using Graph Pad Prism software (San Diego, CA, USA). Student’s t-tests and one-way and two-way analysis of variance were performed using Bonferroni post hoc analyses to determine s.e.m. with a 95% confidence level. Kaplan–Meier survival analyses were analyzed by the log rank test, and comparisons of median disease onset and survival time were analyzed by the Wilcoxon signed rank test.
References
Zinman L, Cudkowicz M . Emerging targets and treatments in amyotrophic lateral sclerosis. Lancet Neurol 2011; 10: 481–490.
van Blitterswijk M, DeJesus-Hernandez M, Rademakers R . How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Curr Opin Neurol 2012; 25: 689–700.
Wijesekera LC, Leigh PN . Amyotrophic lateral sclerosis. Orphanet J Rare Dis 2009; 4: 3.
Brenner D, Muller K, Wieland T, Weydt P, Bohm S, Lule D et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 2016; 139 (Pt 5): e28.
Kenna KP, van Doormaal PT, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet 2016; 48: 1037–1042.
Mueller C, Flotte TR . Clinical gene therapy using recombinant adeno-associated virus vectors. Gene Therapy 2008; 15: 858–863.
Muramatsu S, Fujimoto K, Kato S, Mizukami H, Asari S, Ikeguchi K et al. A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson's disease. Mol Ther 2010 18: 1731–1735.
Marks WJ Jr, Bartus RT, Siffert J, Davis CS, Lozano A, Boulis N et al. Gene delivery of AAV2-neurturin for Parkinson's disease: a double-blind, randomised, controlled trial. Lancet Neurol 2010; 9: 1164–1172.
LeWitt PA, Rezai AR, Leehey MA, Ojemann SG, Flaherty AW, Eskandar EN et al. AAV2-GAD gene therapy for advanced Parkinson's disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol 2011; 10: 309–319.
Christine CW, Starr PA, Larson PS, Eberling JL, Jagust WJ, Hawkins RA et al. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 2009; 73: 1662–1669.
Cearley CN, Wolfe JH . Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol Ther 2006; 13: 528–537.
Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK . Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol 2009; 27: 59–65.
Duque S, Joussemet B, Riviere C, Marais T, Dubreil L, Douar AM et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther 2009; 17: 1187–1196.
Foust KD, Salazar DL, Likhite S, Ferraiuolo L, Ditsworth D, Ilieva H et al. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol Ther 2013; 21: 2148–2159.
Thomsen GM GG, Latter J, Chen M, Vit JP, Staggenborg K, Avalos P et al. Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex. J Neurosci 2014; 34: 15587–15600.
Stoica L, Todeasa SH, Toro Cabrera G, Salameh JS, ElMallah MK, Mueller C et al. AAV delivered artificial microRNA extends survival and delays paralysis in an amyotrophic lateral sclerosis mouse model. Ann Neurol 2016; 79: 687–700.
Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol 2010; 28: 271–274.
Passini MA, Bu J, Richards AM, Treleaven CM, Sullivan JA, O'Riordan CR et al. Translational fidelity of intrathecal delivery of self-complementary AAV9-survival motor neuron 1 for spinal muscular atrophy. Hum Gene Ther 2014; 25: 619–630.
Meyer K, Ferraiuolo L, Schmelzer L, Braun L, McGovern V, Likhite S et al. Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose-response study in mice and nonhuman primates. Mol Ther 2015; 23: 477–487.
Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F . GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993; 260: 1130–1132.
Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M et al. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science 1994; 266: 1062–1064.
Ibanez CF, Andressoo JO . Biology of GDNF and its receptors - relevance for disorders of the central nervous system. Neurobiol Dis 2016; 97: 80–89.
Tovar YRLB, Ramirez-Jarquin UN, Lazo-Gomez R, Tapia R . Trophic factors as modulators of motor neuron physiology and survival: implications for ALS therapy. Front Cell Neurosci 2014; 8: 61.
Rakowicz WP, Staples CS, Milbrandt J, Brunstrom JE, Johnson EM Jr . Glial cell line-derived neurotrophic factor promotes the survival of early postnatal spinal motor neurons in the lateral and medial motor columns in slice culture. J Neurosci 2002; 22: 3953–3962.
Zhang Z, Miyoshi Y, Lapchak PA, Collins F, Hilt D, Lebel C et al. Dose response to intraventricular glial cell line-derived neurotrophic factor administration in Parkinsonian monkeys. J Pharmacol Exp Ther 1997; 282: 1396–1401.
Gash DM, Zhang Z, Ai Y, Grondin R, Coffey R, Gerhardt GA . Trophic factor distribution predicts functional recovery in Parkinsonian monkeys. Ann Neurol 2005; 58: 224–233.
Suzuki M, McHugh J, Tork C, Shelley B, Klein SM, Aebischer P et al. GDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALS. PLoS ONE 2007; 2: e689.
Suzuki M, McHugh J, Tork C, Shelley B, Hayes A, Bellantuono I et al. Direct muscle delivery of GDNF with human mesenchymal stem cells improves motor neuron survival and function in a rat model of familial ALS. Mol Ther 2008; 16: 2002–2010.
Nichols NL, Gowing G, Satriotomo I, Nashold LJ, Dale EA, Suzuki M et al. Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. Am J Respir Crit Care Med 2013; 187: 535–542.
Klein SM, Behrstock S, McHugh J, Hoffmann K, Wallace K, Suzuki M et al. GDNF delivery using human neural progenitor cells in a rat model of ALS. Hum Gene Ther 2005; 16: 509–521.
Wang LJ, Lu YY, Muramatsu S, Ikeguchi K, Fujimoto K, Okada T et al. Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J Neurosci 2002; 22: 6920–6928.
Lu YY, Wang LJ, Muramatsu S, Ikeguchi K, Fujimoto K, Okada T et al. Intramuscular injection of AAV-GDNF results in sustained expression of transgenic GDNF, and its delivery to spinal motoneurons by retrograde transport. Neurosci Res 2003; 45: 33–40.
Acsadi G, Anguelov RA, Yang H, Toth G, Thomas R, Jani A et al. Increased survival and function of SOD1 mice after glial cell-derived neurotrophic factor gene therapy. Hum Gene Ther 2002; 13: 1047–1059.
Manabe Y, Nagano I, Gazi MS, Murakami T, Shiote M, Shoji M et al. Glial cell line-derived neurotrophic factor protein prevents motor neuron loss of transgenic model mice for amyotrophic lateral sclerosis. Neurol Res 2003; 25: 195–200.
Xue YQ, Ma BF, Zhao LR, Tatom JB, Li B, Jiang LX et al. AAV9-mediated erythropoietin gene delivery into the brain protects nigral dopaminergic neurons in a rat model of Parkinson's disease. Gene Therapy 2010; 17: 83–94.
Dufour BD, Smith CA, Clark RL, Walker TR, McBride JL . Intrajugular vein delivery of AAV9-RNAi prevents neuropathological changes and weight loss in Huntington's disease mice. Mol Ther 2014; 22: 797–810.
Manfredsson FP, Tumer N, Erdos B, Landa T, Broxson CS, Sullivan LF et al. Nigrostriatal rAAV-mediated GDNF overexpression induces robust weight loss in a rat model of age-related obesity. Mol Ther 2009; 17: 980–991.
Kells AP, Eberling J, Su X, Pivirotto P, Bringas J, Hadaczek P et al. Regeneration of the MPTP-lesioned dopaminergic system after convection-enhanced delivery of AAV2-GDNF. J Neurosci 2010; 30: 9567–9577.
Su X, Kells AP, Huang EJ, Lee HS, Hadaczek P, Beyer J et al. Safety evaluation of AAV2-GDNF gene transfer into the dopaminergic nigrostriatal pathway in aged and Parkinsonian rhesus monkeys. Hum Gene Ther 2009; 20: 1627–1640.
Kordower JH, Palfi S, Chen EY, Ma SY, Sendera T, Cochran EJ et al. Clinicopathological findings following intraventricular glial-derived neurotrophic factor treatment in a patient with Parkinson's disease. Ann Neurol 1999; 46: 419–424.
Nutt JG, Burchiel KJ, Comella CL, Jankovic J, Lang AE, Laws ER Jr et al. Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology 2003; 60: 69–73.
Gill SS, Patel NK, Hotton GR, O'Sullivan K, McCarter R, Bunnage M et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med 2003; 9: 589–595.
Slevin JT, Gerhardt GA, Smith CD, Gash DM, Kryscio R, Young B . Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J Neurosurg 2005; 102: 216–222.
Lang AE, Gill S, Patel NK, Lozano A, Nutt JG, Penn R et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol 2006; 59: 459–466.
Li W, Brakefield D, Pan Y, Hunter D, Myckatyn TM, Parsadanian A . Muscle-derived but not centrally derived transgene GDNF is neuroprotective in G93A-SOD1 mouse model of ALS. Exp Neurol 2007; 203: 457–471.
Guillot S, Azzouz M, Deglon N, Zurn A, Aebischer P . Local GDNF expression mediated by lentiviral vector protects facial nerve motoneurons but not spinal motoneurons in SOD1(G93A) transgenic mice. Neurobiol Dis 2004; 16: 139–149.
Zhao Z, Alam S, Oppenheim RW, Prevette DM, Evenson A, Parsadanian A . Overexpression of glial cell line-derived neurotrophic factor in the CNS rescues motoneurons from programmed cell death and promotes their long-term survival following axotomy. Exp Neurol 2004; 190: 356–372.
Yang C, Hutto D, Sah DW . Distribution of GDNF family receptor alpha3 and RET in rat and human non-neural tissues. J Mol Histol 2006; 37: 69–77.
Glerup S, Lume M, Olsen D, Nyengaard JR, Vaegter CB, Gustafsen C et al. SorLA controls neurotrophic activity by sorting of GDNF and its receptors GFRalpha1 and RET. Cell Rep 2013; 3: 186–199.
Behrstock S, Ebert A, McHugh J, Vosberg S, Moore J, Schneider B et al. Human neural progenitors deliver glial cell line-derived neurotrophic factor to Parkinsonian rodents and aged primates. Gene Therapy 2006; 13: 379–388.
Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA. 2002; 99: 1604–1609.
Basso DM, Beattie MS, Bresnahan JC . A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 1995; 12: 1–21.
Acknowledgements
We thank Dr Soshana Svendsen for critically reviewing and editing the manuscript, and Drs Brian Kaspar and Vaithi Arumugaswami for providing AAV9-GDNF and AAV9(−), respectively. This work was funded by the ALS Association (ALSA) and the Board of Governors Regenerative Medicine Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Thomsen, G., Alkaslasi, M., Vit, JP. et al. Systemic injection of AAV9-GDNF provides modest functional improvements in the SOD1G93A ALS rat but has adverse side effects. Gene Ther 24, 245–252 (2017). https://doi.org/10.1038/gt.2017.9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2017.9
This article is cited by
-
Bimodal regulation of axonal transport by the GDNF-RET signalling axis in healthy and diseased motor neurons
Cell Death & Disease (2022)
-
Approaches to Gene Modulation Therapy for ALS
Neurotherapeutics (2022)
-
Transplantation of human neural progenitor cells secreting GDNF into the spinal cord of patients with ALS: a phase 1/2a trial
Nature Medicine (2022)
-
Current knowledge and challenges associated with targeted delivery of neurotrophic factors into the central nervous system: focus on available approaches
Cell & Bioscience (2021)
-
Specific Expression of Glial-Derived Neurotrophic Factor in Muscles as Gene Therapy Strategy for Amyotrophic Lateral Sclerosis
Neurotherapeutics (2021)
