
Abstract
Purpose: To use the Hunter Outcome Survey, an international database, to assess the safety and effectiveness of enzyme replacement therapy with idursulfase in patients with Hunter syndrome who started treatment before 6 years of age.
Methods: The study population included all patients enrolled in the Hunter Outcome Survey who started idursulfase infusions (0.5 mg/kg every other week) before 6 years of age and who had at least one follow-up examination recorded.
Results: The study population included 124 patients, younger than 6 years, who had a mean age at start of idursulfase of 3.6 ± 1.6 years (mean ± SD). The mean duration of treatment was 22.9 ± 14.6 months. A total of 69 infusion-related reactions occurred in 33 (26.6%) patients, including three serious infusion-related reactions occurring in a single patient. After at least 6 months of idursulfase, urine glycosaminoglycan levels decreased from 592 ± 188 to 218 ± 115 μg/mg creatinine (P < 0.0001, n = 34). Liver size, estimated by palpation, was also significantly decreased (P = 0.005, n = 23). Similar safety and effectiveness results were seen in patients who were aged 6 years or older when initiating idursulfase.
Conclusion: No new safety concerns were identified in patients younger than 6 years, and clinical benefit was suggested by the reduction in liver size.
Similar content being viewed by others

Main
Hunter syndrome (mucopolysaccharidosis type II [MPS II, OMIM# 309900]) is an X-linked disorder caused by a deficiency in the lysosomal enzyme, iduronate-2-sulfatase.1,2 This enzyme deficiency results in the cellular accumulation of partially degraded glycosaminoglycan (GAG) fragments from dermatan sulfate and heparan sulfate,1 causing multisystem disease. The incidence of Hunter syndrome is estimated to be approximately 1 in 162,000 live male births.3
Patients with Hunter syndrome typically appear normal at birth, with signs and symptoms emerging between 2 and 4 years of age. The initial clinical features include coarse facial features, enlarged tongue, tonsils, and adenoids resulting in upper respiratory obstructions and frequent ear infections.4–6 Hepatomegaly, splenomegaly, joint stiffness and skeletal abnormalities, and cardiac disease are common.1,7 Although a wide spectrum of clinical severity is observed, two phenotypes are recognized—severe and attenuated. Patients with either phenotype exhibit the somatic signs and symptoms listed earlier. Additionally, patients with the severe phenotype demonstrate central neurologic involvement resulting in profound cognitive impairment and developmental regression.6 In the severely affected patients, death typically occurs in the first or second decade of life,1 whereas patients with the attenuated phenotype typically survive into adulthood, although premature mortality occurs.
Enzyme replacement therapy (ERT) with recombinant human iduronate-2-sulfatase (idursulfase, Elaprase®, Shire Human Genetics Therapies, Inc., Cambridge, MA) has been commercially available since 20068 and has been associated with clinically relevant reductions in spleen and liver volume in clinical trials,8,9 as well as with significant improvement in a composite measure of pulmonary function and mobility.8 The patients in these clinical trials were all at least 5 years of age and had substantial somatic involvement at baseline. Initiation of ERT may slow or stop the progression of the pathologies of Hunter syndrome before irreversible changes occur, but little data exist to support the safety or effectiveness of early intervention. In this study, we describe results from the Hunter Outcome Survey (HOS) on the safety and effectiveness of idursulfase in patients younger than 6 years and compare these findings with those seen in the older patients.
METHODS
Hunter Outcome Survey
HOS is an international, long-term observational survey designed to increase the knowledge of the natural history of Hunter syndrome and to evaluate the safety and effectiveness of ERT with idursulfase. All participating centers received approval according to local regulations, which are typically those of an Institutional Review Board or Ethics Committee, before enrolling any patients into HOS. HOS is controlled by physicians and is overseen by national, regional, and international advisory boards comprising physicians experienced in the management and treatment of patients with Hunter syndrome. Enrollment into HOS began in October 2005. Data in HOS were collected during examination of patients as part of routine medical care. A description of the data collected in HOS from participating physicians and the mechanics of data recording, transfer, and analysis have been published previously.7
Patients
Any patient with a biochemically or genetically confirmed diagnosis of Hunter syndrome is eligible to enroll in HOS. Before enrolling in HOS, all patients, their parents, or their legal representative provided informed, written consent for participation. The study population for this analysis included all enrolled patients who were treated with ERT and who had at least one follow-up examination recorded in HOS as of April 23, 2010.
Study design
HOS is an observational database designed to collect the natural history and long-term safety and effectiveness data of idursulfase in MPS II patients. For patients who began ERT before entry into HOS, adverse events were recorded only after entry into HOS. Infusion-related reactions (IRRs), which typically involve pyrexia, rigors, and flushing, were analyzed separately and were defined as events occurring during or within 24 hours of an infusion that had evidence of a causal relationship with idursulfase. Urine GAG levels were determined by methods used in the local or central laboratories. Liver size was estimated by palpation. Frozen plasma samples were transported to Shire HGT where antiidursulfase antibodies were detected by an enzyme-linked immunosorbent assay as described previously.8
Statistical analysis
All statistical analysis was conducted by Shire Human Genetic Therapies under the direction of the HOS advisory boards. The results are presented using descriptive statistics including prevalence (i.e., n [%]), mean, median, standard deviation, and percentiles. Results are generally presented as mean ± standard deviation. Because of the nonnormal distribution of some data, the median was used to accurately summarize the results of some variables. Height, weight, and head circumference of HOS participants were compared with age-specific measurements for the general pediatric population. All analyses were done using SAS software (SAS Institute, Cary, NC).
RESULTS
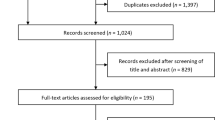
A total of 644 prospective patients (i.e., patients who were alive at entry into HOS) had enrolled in HOS as of April 23, 2010. A total of 461 patients had received ERT, and 145 patients had started ERT before reaching the age of 6 years (Fig. 1). Of these 145 patients younger than 6 years, 124 had at least one follow-up visit in HOS and comprised the study population. Figure 2 presents the age at initiation of ERT of the population <6 years of age in four geographical regions. These 124 patients were treated in 54 clinics located in 18 countries in Europe, North America, Latin America, and Asia. The characteristics of this patient population (<6 years of age) on entry into HOS are presented in Table 1, where they are compared with the enrolled patients 6 years or older when they started ERT and with all prospective patients enrolled in HOS. The average delay between the emergence of symptoms and the diagnosis of Hunter syndrome in patients younger than 6 years was 0.9 ± 1.3 years (mean ± SD, median = 0.8 years), when compared with 2.2 ± 2.8 years (median = 1.5 years) for patients 6 years or older (Table 1).

The age distribution of ERT-treated patients in HOS. ERT, enzyme replacement therapy; HOS, Hunter Outcome Survey.

Distribution of the age at initiation of ERT in patients with Hunter syndrome younger than 6 years in Europe, North America, Latin America, and Asia.
In the study population of patients younger than 6 years, the mean age at start of ERT was 3.6 ± 1.6 years. Eleven patients were younger than 12 months at the start of ERT (Fig. 2). The duration of treatment from HOS entry to last visit was 20.0 ± 10.6 months. Seventy of the patients started treatment with idursulfase before HOS entry. When the duration of ERT before HOS entry was included, the mean time of ERT at the time of last study visit was 22.9 ± 14.6 months. The estimated mean weekly dose of idursulfase, after HOS entry, was 0.53 mg/kg (median = 0.50 mg/kg, 10th–90th percentile = 0.45–0.63 mg/kg). For the majority of patients in the <6-year-old study population (91 of 106 patients with height data recorded in HOS), length or height was within the normal range for age10 (data not shown). In contrast, 104 of 117 patients had body weight above the 50th percentile of the normal range for age, and 37 of 117 patients had a body weight exceeding the 97th percentile of the normal range for age (data not shown).
Table 2 presents the prevalence of neurologic involvement in ERT-treated children younger than 6 years and children 6 years or older who had available data. Incidence of behavioral and cognitive problems and hyperactivity was similar between the two groups. However, specific reported incidences of carpal tunnel syndrome, gait problems, history of seizures, difficulty swallowing, and deficits in fine motor skills were seen more often in patients 6 years or older. Table 3 further compares the characteristics of children with Hunter syndrome younger than 6 years with children 6 years or older in their developmental milestones and cognitive achievements.
Safety
A total of 69 nonserious IRRs were reported in 33 (26.6%) patients younger than 6 years (Table 4). Three serious IRRs were reported in a single patient in the <6-year-old patient subgroup. These serious IRRs included one cough and bronchospasm event and urticaria on two occasions. In response to these serious IRRs, the infusion was stopped, and the patient was treated with betamethasone. All but two of the nonserious IRRs were classified as mild or moderate severity. One of the episodes of bronchospasm and one IRR noted earlier were classified as severe, and both resolved without sequelae.
In patients 6 years or older, a total of 104 nonserious IRRs were reported in 50 (17.4%) patients. Six patients (2.1%) in the ≥6-year-old group had a serious IRR, including two episodes of urticaria, two IRRs classified as general disorders, one rash, and one decrease in blood pressure. Two of these IRRs were classified as moderate severity, three were classified as severe, and one missing. In the three IRRs classified as severe, the infusions were stopped, and patients were given medication and were observed in the hospital overnight.
In the <6-year-old group, a total of 28 serious adverse events were reported in 16 patients (12.9%). These events included 10 planned surgeries, including release of carpal tunnel (n = 2), adenoidectomy and/or tonsillectomy (n = 2), repair of inguinal hernia (n = 1), placement or replacement of a venous access device (n = 4), and tooth extraction (n = 1). One patient was hospitalized with bilateral subdural hematoma, evacuated through a burr hole. No further surgical intervention was performed. A routine echocardiogram in another patient revealed the presence of a 2-cm clot in the right atrium that was attached to an indwelling central catheter and the atrial wall. The patient was placed on anticoagulant therapy and discharged from the hospital.
Additional serious adverse events included two hospitalizations for replacement of an infected venous access device (n = 1), replacement of a malfunctioning venous access device (n = 1), and the three IRRs described earlier. The three serious IRRs were classified as probably (n = 2) and possibly (n = 1) drug- related. None of the other serious adverse events was classified as possibly or probably related to idursulfase.
In the ≥6-year-old group, a total of 92 serious adverse events were reported in HOS for 58 patients (20.2%). These events included 20 infections, 9 surgical and medical procedures, 5 reports of respiratory failure, 3 episodes of respiratory distress, 2 reports of obstructive airway disorder, 2 IRRs noted above, and 5 deaths. Seven of these serious adverse events were considered to be possibly (n = 1) or probably (n = 6) related to drug treatment. All adverse events that were classified as possibly or probably drug-related occurred in association with in-hospital idursulfase infusions.
Antibodies
No IgE antiidursulfase antibodies were detected in the plasma from any patients. In patients younger than 6 years, IgG antiidursulfase antibodies were detected in 38 of 71 (53.5%) patients whose plasma was assayed. Seventy-one of 166 (42.8%) patients 6 years or older demonstrated IgG antiidursulfase antibodies.
Effectiveness
Urine GAG levels significantly decreased from baseline to postbaseline in the 40 patients younger than 6 years, who had data available for urine GAG levels both before and after at least 6 months of idursulfase (Fig. 3). Patients 6 years or older also experienced a reduction in urine GAG levels (n = 84, Fig. 3). In the subgroup of 34 patients younger than 6 years with elevated baseline GAG levels (>200 μg/mg creatinine), mean urine GAG levels decreased from 592 ± 188 μg/mg creatinine at baseline to 218 ± 115 μg/mg creatinine after at least 6 months of idursulfase (P < 0.0001, Wilcoxon signed rank test). In the subgroup of 43 patients 6 years or older with elevated GAG levels, mean urine GAG levels decreased from 363 ± 111 μg/mg creatinine at baseline to 125 ± 77 μg/mg creatine after at least 6 months (P < 0.0001, Wilcoxon signed rank test).

Urine GAG levels before and after at least 6 months of idursulfase. GAG, glycosaminoglycan; ERT, enzyme replacement therapy. Baseline refers to the closest measurement within 12 months before starting ERT, and postbaseline refers to the first measurement made at least 6 months after starting ERT. The box plots show 10th, 25th, 75th, and 90th percentile and the median. The filled squares represent the mean values. The P value from the Wilcoxon signed-rank test is P < 0.0001 for both patient groups.
Liver size estimated by palpation was larger than normal at baseline in both groups of patients, with the size being larger in the older patients compared with the younger patients (Fig. 4). As shown in Figure 4, after at least 6 months of idursulfase, liver size was significantly decreased in both groups.

Liver size before and after at least 6 months of idursulfase. Baseline refers to the closest measurement within 12 months before starting ERT, and postbaseline refers to the first measurement made at least 6 months after starting ERT. The box plots show 10th, 25th, 75th, and 90th percentile and the median. The filled squares represent the mean values. The P value from the Wilcoxon signed-rank test is P = 0.005 for the <6-year-old group and P < 0.0001 for the ≥6-year-old group.
DISCUSSION
This analysis of patients in HOS who began treatment with idursulfase at <6 years of age compared with the patients who started idursulfase after 6 years of age revealed no new or unanticipated safety concerns. Urine GAG levels were reduced 63% in patients younger than 6 years, which is similar to the reduction (66%) in HOS patients older than 6 years. Urine GAG levels were reduced 52.5% after 1 year of weekly idursulfase (0.5 mg/kg) infusions in the Phase II/III ERT clinical trial that resulted in idursulfase approval.8 The baseline data highlight the clinical observations that urine GAG levels are higher in young patients with Hunter syndrome compared with older patients. The etiology of the lower urine GAG levels in older patients with Hunter syndrome and normal adults compared with normal young children is unknown. The significant reduction in urine GAG indicates that idursulfase is biologically active in patients younger than 6 years, and the reduction in liver size after at least 6 months of treatment is suggestive of a clinical benefit.
Hunter syndrome is a progressive disease. Patients typically appear normal at birth, and clinical features begin to emerge between the ages of 2 and 4 years. Although no longitudinal natural history studies have been published, cross-sectional studies clearly show substantial somatic involvement before the age of 6 years, including skeletal and joint abnormalities.5–7 Of interest in this study was the apparent shorter time between onset of symptoms and diagnosis of Hunter syndrome in patients younger than 6 years compared with patients 6 years or older (Table 1), although considerable overlap in delay in diagnosis was seen. The cause of this difference is unknown but may reflect a recent heightened awareness of Hunter syndrome after the approval of ERT for Hunter syndrome.
The goal of ERT in Hunter syndrome is to slow or stop the progression of the disease, and thus, it is important to initiate treatment before the onset of irreversible changes. The issue of at what age or at what stage of the disease progression to initiate ERT has not been established. The results of this study support the use of idursulfase in patients younger than 6 years as no new safety issues were seen compared with the experience in older patients.8,9 However, the issue of proving clinical benefit in young patients remains problematic, because functional testing requires the cooperation of the patient, particularly when assessing pulmonary function or mobility. Although the reduction in liver size seen in this study may indicate a clinical benefit, long-term follow-up will be necessary to determine whether early initiation of ERT slows the progression of the disease sufficiently to result in clinical benefit.
Approximately 75% of patients with Hunter syndrome are destined to express the severe phenotype,11 but the prediction of an attenuated or severe phenotype is difficult early in the course of the disease. However, by the age of 5 or 6 years, the phenotype should be clear. Thus, this study included patients with both phenotypes, because most of the children in this study were too young for it to be determined whether they would ultimately express the severe phenotype. Intravenous idursulfase is not expected to cross the blood-brain barrier in sufficient quantity to influence the central neurologic involvement in these patients. However, patients with the severe phenotype may expect somatic benefits of ERT.
IRRs were the most common drug-associated adverse event observed in these two subpopulations in HOS. The incidence, but not the severity, of all IRRs, seems to be higher in the <6-year-old subpopulation than in the ≥6-year-old group (26.6% versus 18.8% of patients, respectively). In the Phase II/III clinical trial of idursulfase,8 22 of 32 (68.8%) patients (age, 5–33 years) treated with idursulfase 0.5 mg/kg weekly experienced one or more IRRs. In that study, the incidence of IRRs was greatest between treatment weeks 4 and 12 and decreased thereafter.8 In this study, the incidence of IRRs was considerably less in both age groups than has been reported previously in clinical trials of idursulfase. IRRs are typically managed by slowing the infusion rate and/or administration of an antihistamine. Patients who have previously experienced an IRR are premedicated with an antihistamine and/or corticosteroids before subsequent infusions, and most can be weaned from premedications after demonstration of incident-free treatment.8
Exogenous idursulfase could be viewed as “foreign” by many patients with Hunter syndrome, and therefore, an immune response may be expected. The IgG antibody response in this study was similar to that reported for the pivotal study of idursulfase.8 In that study, 46.9% of patients treated with weekly or every other week idursulfase (0.5 mg/kg) developed IgG antiidursulfase antibodies. The reduction in urine GAG excretion was attenuated in IgG-positive patients in that study, but no other effects on clinical responses or safety were reported. Antibodies have been reported during ERT for other lysosomal storage diseases, for example, Fabry disease,12–14 MPS VI,15 and MPS I.16 Similarly, IgG antibodies directed against these other exogenous lysosomal enzymes have been associated with attenuation of biochemical responses (e.g., reduction in urine globotriaosylceramide levels in Fabry disease12), but antibody incidence or titer does not seem to be associated with clinical responses (e.g., rate of loss of glomerular filtration rate in Fabry disease).12,17 Further research will be needed to study the impact of antiidursulfase antibodies on the safety and efficacy of ERT in Hunter syndrome.
Idursulfase has been commercially available since 2006, and the published information about its safety and effectiveness is confined to three clinical trials8,9,18 and a small number of case reports (for example, Refs. 19 and 20). HOS is an important resource for evaluating the safety and effectiveness of idursulfase in the “real world” environment. As more patients are enrolled in HOS and more data are acquired about the early initiation of ERT, a consensus on the benefits of early idursulfase use should emerge.
Study limitations
The study was a retrospective analysis of open-labeled treatment of patients in their usual medical environment. The lack of a concurrently followed up untreated or placebo-treated group limits the strength of the observations.
CONCLUSIONS
Idursulfase has been prescribed to many patients with Hunter syndrome younger than 6 years around the world as documented in the present report. No new safety issues have been observed in these young patients treated with idursulfase. Clinical benefit was suggested by the reduction in liver volume of treated patients, but long-term observation will be required to determine whether early initiation can prevent progression of clinical disease in Hunter syndrome.
REFERENCES
Neufeld EF, Muenzer J . The mucopolysaccharidoses. In: Scriver CR, editor. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2001; 3421–3452.
Bach G, Eisenberg F Jr, Cantz M, Neufeld EF . The defect in the Hunter syndrome: deficiency of sulfoiduronate sulfatase. Proc Natl Acad Sci USA 1973; 70: 2134–2138.
Meikle PJ, Hopwood JJ, Clague AE, Carey WF . Prevalence of lysosomal storage disorders. JAMA 1999; 281: 249–254.
Martin R, Beck M, Eng C, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 2008; 121: e377–e386.
Young ID, Harper PS . Mild form of Hunter's syndrome: clinical delineation based on 31 cases. Arch Dis Child 1982; 57: 828–836.
Young ID, Harper PS . The natural history of the severe form of Hunter's syndrome: a study based on 52 cases. Dev Med Child Neurol 1983; 25: 481–489.
Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J . Initial report from the Hunter Outcome Survey (HOS). Genet Med 2008; 10: 508–516.
Muenzer J, Wraith JE, Beck M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med 2006; 8: 465–473.
Muenzer J, Gucsavas-Calikoglu M, McCandless S, Schuetz T, Kimura A . A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol Genet Metab 2007; 90: 329–337.
Kuczmarski RJ, Ogden CL, Guo SS, et al. 2000 CDC Growth Charts for the United States: methods and development. Vital Health Stat 11 2002; 1–190.
Young ID, Harper PS, Newcombe RG, Archer IM . A clinical and genetic study of Hunter's syndrome. 2. Differences between the mild and severe forms. J Med Genet 1982; 19: 408–411.
Schiffmann R, Ries M, Timmons M, Flaherty JT, Brady RO . Long-term therapy with agalsidase alfa for Fabry disease: safety and effects on renal function in a home infusion setting. Nephrol Dial Transplant 2006; 21: 345–354.
Schiffmann R, Kopp JB, Austin HA, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 2001; 285: 2743–2749.
Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human α-galactosidase A-replacement therapy in Fabry's disease. N Engl J Med 2001; 345: 9–16.
Harmatz P, Giugliani R, Schwartz I, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J Pediatr 2006; 148: 533–539.
Wraith JE, Clarke LA, Beck M, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr 2004; 144: 581–588.
Benichou B, Goyal S, Sung C, Norfleet AM, O'Brien F . A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol Genet Metab 2009; 96: 4–12.
Okuyama T, Tanaka A, Suzuki Y, et al. Japan Elaprase Treatment (JET) study: idursulfase enzyme replacement therapy in adult patients with attenuated Hunter syndrome (mucopolysaccharidosis II, MPS II). Mol Genet Metab 2010; 99: 18–25.
Little C, Gould R, Hendriksz C . The management of children with Hunter syndrome—a case study. Br J Nursing 2009; 18: 321–322.
Galan-Gomez E, Guerrero-Rico A, Caceres-Marzal C, et al. Early response to idursulfase treatment in a 3 year-old boy affected of Hunter syndrome. Eur J Med Genet 2008; 51: 268–271.
Acknowledgements
Supported by the NIHR Manchester Biomedical Research Centre (J.E.W.). The authors thank all the patients who have participated in HOS, their parents and guardians, and their treating physicians and staff. In addition, the authors thank Ms. Kathryn Harnett of Shire HGT for her excellent statistical support during the preparation of this report. Editorial assistance to the authors was provided by Edward Weselcouch, PhD, of PharmaWrite (Princeton, NJ) and was paid for by Shire HGT. The authors participated fully in the preparation of this manuscript and are fully responsible for its content.
The following physicians and clinics have contributed data to HOS:
Argentina—Buenos Aires: Hernan Amartino and Cecilia Riccheri and Cordoba: Norberto Guelbert. Austria—Graz: Barbara Plecko and Salzburg: Olaf Bodamer. Belgium—Brussels: Linda De Meirleir. Brazil—Fortaleza: Erlane Ribeiro; Maceio: Emerson Santos; Porto Alegre: Roberto Giugliani; Rio de Janeiro: Raquel Tavares Boy da Silva; Salvador: Angelina Acosta; Sao Paulo: Ana Martins. Bulgaria—Sofia: Radka Tincheva. Canada—Calgary: Robin Casey; Edmonton: Alicia Chan; Toronto: Joe Clarke and Julian Raiman; Vancouver: Lorne Clarke. Croatia—Zagreb: Ingeborg Barišić and Ivo Barić. Czech Republic—Prague: Jiri Zeman. Denmark—Copenhagen: Allan Meldgaard Lund. France—Lyon: Nathalie Guffon; Paris: Vassili Valayannopoulos and Bénédicte Héron. Germany—Berlin: Julia Hennermann; Hamburg: Nicole Muschol; Magdeburg: Silke Klose; Mainz: Michael Beck, Christoph Kampmann, and Christina Lampe. Greece—Thessaloniki: Dimitrios Zafeiriou. Italy—Ancona: Orazio Gabrielli; Bari: Francesco Papadia; Bologna: Alessandro Cicognani; Catania: Fiumara Rita; Catanzaro: Daniela Concolino; Firenze: Alinic Donati; Genova: Maja Di Rocco; Monza: Rossella Parini; Napoli: Andria Generoso; Padova: Maurizio Scarpa; Rome: Claudio Feliciani. Portugal—Lisbon: Ana Gaspar; Porto: Elisa Leao Teles and Esmeralda Martins. Russia—Moscow: Peter Novikov. Spain—Almeria: Javier Aguirre; Badajoz: Enrique Galán; Badalona: Guillem Pintos; Barcelona: Mireia del Toro and Merce Pined; Bilbao: Luis Aldamiz; Huelva: Olga Alonso; Las Palmas: Milagros Marti; Linares: Pilar Munguira; Madrid: Luis González Gutiérrez-Solana; Murcia: Rosario Domingo and Encarna Guillen; Ourense: Gemma Novoa; Palma de Mallorca: Begoña de Azua; Salamanca: Aránzazu Hernández; Seville: Dolores Lluch and Jose Luiz Diaz; Valencia: Jaime Dalmau; Valladolid: José Manuel Muro and Carlos Alcalde; Zaragoza: Juan Pérez, Immaculada Garcia, and Marta Ferrer. Sweden—Gothenburg: Niklas Darin; Halmstad: Nils O Nilsson; Lund: Dominiki Papadopoulou; Stockholm: Gunilla Malm and Ingrid Dahlman. Taiwan—Taipei: Shuan-Pei Lin. The Netherlands—Rotterdam: Ans van der Ploeg. The United Kingdom—Amersham: UK MPS Society; Birmingham: Chris Hendriksz; Cambridge: Uma Ramaswami; London: Ashok Vellodi; Manchester: Simon Jones, James Edmond Wraith, and Stephen Waldek. The United States—Atlanta, GA: Paul Fernhoff; Baltimore, MD: Ada Hamosh; Boston, MA: Katherine Sims; Chapel Hill, NC: Joseph Muenzer; Charlottesville, VA: William Wilson; Chicago, IL: Barbara Burton; Cincinnati, OH: Nancy Leslie; Columbus, OH: Kim McBride; Denver, CO: Janet Thomas; Greenville, SC: Curtis Rogers; Hartford, CT: Robert Greenstein; Houston, TX: Christine Eng; Iowa City, IA: Val Sheffield; Jackson, MS: Omar Abdul-Rahman; Kansas City, MO: Laurie Smith; Miami, FL: Parul Jayakar; Minneapolis, MN: Nancy Mendelsohn, Chet Whitley; New York, NY: Greg Pastores; Norfolk, VA: Virginia Proud; Oakland, CA: Paul Harmatz; Omaha, NE: William Rizzo; Orange, CA: Raymond Wong; Paterson, NJ: Jennifer Ibrahim; Phoenix, AZ: Kirk Aleck; Portland, OR: Robert Steiner; St. Louis, MO: Dorothy Grange; Salt Lake City, UT: David Viskochil; Seattle, WA: Ronald Scott; Sioux Falls, SD: Laura Keppen; Washington, DC: Cynthia Tifft.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: Dr. Muenzer has received honoraria, travel grants, and/or research grants from Shire HGT and Genzyme Corporation. Dr. Beck has received research funding, consultancy fees, and/or speaker's fees from Shire HGT, Genzyme Corporation, Actelion Pharmaceuticals Ltd, and BioMarin. Dr. Giugliani has received consultancy fees, travel grants, and/or speaker's fees from the following companies: Shire HGT, Genzyme Corporation, BioMarin, Actelion Pharmaceuticals Ltd, and Amicus. Dr. Suzuki has received research funding, consultancy fees, and/or speaker's fees from Genzyme Corporation. Dr. Tylki-Szymanska has received consultancy and speaker's fees from Shire HGT and Genzyme Corporation. Dr. Valayannopoulos is a member of the Hunter Advisory Board sponsored by Shire HGT and has received consultancy fees from Shire HGT. Dr. Vellodi reports receiving honoraria, research support, and/or travel grants from Shire HGT. Dr. Wraith has received research funding, consultancy fees, and/or speaker's fees from Shire HGT, Genzyme Corporation, Actelion Pharmaceuticals Ltd, and BioMarin.
Rights and permissions
About this article
Cite this article
Muenzer, J., Beck, M., Giugliani, R. et al. Idursulfase treatment of Hunter syndrome in children younger than 6 years: Results from the Hunter Outcome Survey. Genet Med 13, 102–109 (2011). https://doi.org/10.1097/GIM.0b013e318206786f
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e318206786f
Keywords
This article is cited by
-
Transabdominal preperitoneal repair for an adolescent patient with Hunter syndrome: a case report
Surgical Case Reports (2019)
-
Ten years of the Hunter Outcome Survey (HOS): insights, achievements, and lessons learned from a global patient registry
Orphanet Journal of Rare Diseases (2017)
-
Clinical outcomes in idursulfase-treated patients with mucopolysaccharidosis type II: 3-year data from the hunter outcome survey (HOS)
Orphanet Journal of Rare Diseases (2017)
-
The impact of the immune system on the safety and efficiency of enzyme replacement therapy in lysosomal storage disorders
Journal of Inherited Metabolic Disease (2016)
-
Effect of systemic high dose enzyme replacement therapy on the improvement of CNS defects in a mouse model of mucopolysaccharidosis type II
Orphanet Journal of Rare Diseases (2015)
