
Abstract
Purpose:
Genomic sequencing (GS) for newborns may enable detection of conditions for which early knowledge can improve health outcomes. One of the major challenges hindering its broader application is the time it takes to assess the clinical relevance of detected variants and the genes they impact so that disease risk is reported appropriately.
Methods:
To facilitate rapid interpretation of GS results in newborns, we curated a catalog of genes with putative pediatric relevance for their validity based on the ClinGen clinical validity classification framework criteria, age of onset, penetrance, and mode of inheritance through systematic evaluation of published evidence. Based on these attributes, we classified genes to guide the return of results in the BabySeq Project, a randomized, controlled trial exploring the use of newborn GS (nGS), and used our curated list for the first 15 newborns sequenced in this project.
Results:
Here, we present our curated list for 1,514 gene–disease associations. Overall, 954 genes met our criteria for return in nGS. This reference list eliminated manual assessment for 41% of rare variants identified in 15 newborns.
Conclusion:
Our list provides a resource that can assist in guiding the interpretive scope of clinical GS for newborns and potentially other populations.
Genet Med advance online publication 12 January 2017
Similar content being viewed by others
Introduction
Exome or genome sequencing, collectively referred to as genomic sequencing (GS), provides unparalleled opportunities to screen thousands of disorders in newborns that have previously been considered impossible to detect. In addition to identifying risks for treatable childhood-onset diseases, GS may also allow early intervention to improve the outcome for many other genetic disorders, avoid the diagnostic odyssey in ill newborns, make genomic data available for future indications, provide pharmacogenomic information for guiding drug use, and help in reproductive planning. Although there are many benefits, concerns such as identification of variants of uncertain significance and social and economic implications of the findings need to be addressed before GS is more widely adopted.
Another major challenge restricting the wider application of GS is interpreting the vast amount of genomic data within a short timeframe. To ensure that only results of high predictive value are returned, best practice today calls for thorough review of evidence for each variant.1 Laboratories often use bioinformatics tools to reduce the number of variants requiring manual assessment based on a set of filtration criteria such as allele frequency, predicted protein impact, and reported pathogenicity claims. However, an important part of the interpretation process—determining whether the gene impacted by the variant is strongly associated with disease and meets criteria to be returned—involves manually reviewing the validity of each gene’s role in disease and assessing the utility of returning the result using attributes such as penetrance and age of onset. At present, there is no resource of genes curated for these attributes, making analysis laborious and time-consuming. Although it is currently not feasible to predefine all disease-causing variants in advance, it is possible to curate disease-associated genes and predetermine which are appropriate to be reported in a newborn GS (nGS) scenario. Creating and sharing a list of nGS target genes would help accelerate and standardize the interpretation process and facilitate the use of this technology to support newborn screening (NBS) programs.
The BabySeq Project is a randomized, controlled trial to explore medical, behavioral, and economic outcomes associated with the use of GS in newborns. To facilitate nGS results interpretation, we curated 1,514 genes for evidence supporting the gene’s role in disease, age of onset, penetrance, and mode of inheritance based on a set of criteria determined by our interdisciplinary group of clinical and molecular geneticists, pediatricians, neonatologists, and biomedical ethicists. Using this information, we classified each gene as to whether it met the criteria to be returned in nGS. To provide proof of principle that our curated gene–disease association reference list facilitates results interpretation in nGS, we utilized it in the analysis of the first 15 newborns sequenced in the BabySeq Project.
Materials and Methods
The BabySeq project
Two cohorts of newborns and their parents were enrolled in the BabySeq Project: (i) healthy newborns from Brigham and Women’s Hospital Well Newborn Nursery and (ii) ill newborns from Boston Children’s Hospital’s neonatal intensive care unit. Family histories were obtained for each participant at enrollment in a genetic counseling session. Half of the newborns in each cohort were randomized to receive standard care and genetic counseling based on their family histories only; the others received GS in addition to standard care and genetic counseling based on both their GS results and family histories. GS reports of those randomized to receive sequencing were entered into the newborn’s medical record. The impact of GS on newborn clinical care, parent and physician behaviors, and economic outcomes were evaluated in parents using baseline, 3-month, and 10-month postdisclosure surveys and in clinicians using baseline, postdisclosure (GS arm only), and end-of-study surveys. This study was approved by the Boston Children’s Hospital and Partners institutional review boards. Informed consent was obtained from each participant.
Generation of a gene–disease association reference list
Each specific gene–disease pair was curated for the following attributes:
Validity of gene–disease association. Evidence that the gene has a causal role in disease was determined based on the framework released by the Clinical Genome Resource (ClinGen) Gene Curation Working Group (https://www.clinicalgenome.org/working-groups/gene-curation/projects-initiatives/clinical-validity-classifications/). In this method, the validity of a gene–disease association is evaluated by reviewing the evidence reported in the literature, such as the number of families with pathogenic variants in the gene and functional studies, and classified into the following categories: conflicting evidence, no reported evidence, limited evidence, moderate evidence, strong evidence, and definitive evidence.
Age of onset. The youngest age at which individuals with pathogenic variants in the gene presented with disease was curated based on available information in the literature and classified into the following categories: ≤2 years of age, 2–10 years of age, 10–18 years of age, and >18 years of age.
Penetrance. Estimated penetrance was curated based on the phenotype information for reported individuals in the literature and classified as “high” if ≥80% of individuals were symptomatic, “moderate” if 20–80% of individuals were symptomatic, and “low” if <20% of individuals were symptomatic. Because our assertions regarding penetrance were based on the literature without direct phenotyping of individuals, our approach was limited by the number of reported individuals and the phenotype description for affected and control individuals; therefore, our classifications reflect an estimate based on the available literature. To reflect the amount of evidence, a confidence rank was added to each assertion if the assertion was made for (i) genes with definitive or strong evidence and a large number of families or (ii) genes with only a small number of families (moderate evidence–level genes). For genes with limited or conflicting evidence, penetrance was not assessed and was noted as “unknown.”
Inheritance. The most common inheritance pattern for the gene was determined.
Sequencing and results analyses
Sequencing and results analyses were performed as described in the Supplementary Materials and Methods online. Variants were assessed and classified as described.2,3
Results
Establishing criteria for the return of results in nGS
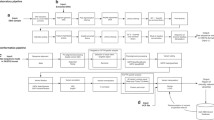
Our interdisciplinary group determined a set of criteria for return of results in nGS that were incorporated into our BabySeq Project protocol. Two distinct reporting strategies were created for the two cohorts in our study in the context of returning results for screening purposes versus for diagnostic testing ( Figure 1a ). A newborn genomic sequencing report (NGSR) was developed to return results relevant to both healthy and ill newborns. The criteria were developed to maximize benefit while minimizing uncertainty from reporting disorders with low penetrance, late onset, or suboptimal evidence for association. The NGSR was restricted to four groups of results: (i) childhood-onset (earliest reported onset before the age of 18) disease risk, involving genes with at least strong evidence to cause highly penetrant childhood-onset disorders; (ii) genes with moderate evidence and/or moderate penetrance associated with conditions for which action during childhood may prevent a devastating outcome later in life, considering that the benefit of learning that a newborn has a pathogenic variant in such a gene is likely to outweigh the uncertainty in disease risk; (iii) genes with strong pharmacogenomic associations (class 1 and 2A genes in the PharmGKB database (https://www.pharmgkb.org)) that are relevant to the pediatric population, including RYR1 associated with malignant hyperthermia, TPMT associated with thiopurine toxicity, and G6PD associated with hemolytic anemia due to glucose-6-phosphate dehydrogenase deficiency; and (iv) carrier status for any gene meeting these criteria ( Figure 1b , c ). Only pathogenic and likely pathogenic variants in these genes were included in the NGSR.

Return of results criteria in the BabySeq project. (a) All newborns in the sequencing group receive a newborn genomic sequencing report (NGSR) that returns risk and carrier status for childhood-onset disease and pharmacogenomics variants that may be relevant to the pediatric population. In addition, sick newborns receive an indication-based analysis (IBA) that returns all variants with evidence to cause or contribute to the infant’s disease, with an option to query pharmacogenomics variants related to the infant’s care. (b) Criteria for genes to be included in the NGSR and IBA. NGSR was limited to genes with strong evidence to cause highly penetrant childhood-onset disorders; while genes related to the infant’s clinical features with moderate evidence or moderate penetrance or typically present at later ages were also included in IBA. When a specific disease is suspected based on the infant’s presentation, genes associated with that disease with limited evidence or low penetrance may also be returned. (c) Criteria for variants to be included in the NGSR and IBA. Only pathogenic and likely pathogenic variants were returned in the NGSR, whereas IBA also included variants of uncertain significance in genes associated with the infant’s indication.
Although only variants classified as pathogenic or likely pathogenic are returned to healthy infants, our group determined that all variants with evidence to support a clear or possible contribution to the infant’s indication should be returned for ill newborns, consistent with professional guidelines.4 Hence, variants of uncertain significance in genes relevant to the newborn’s clinical indication, even if the gene has moderate or limited evidence to cause the specific indication, are also returned to provide an opportunity for follow-up studies that may help clarify their clinical significance ( Figure 1 ).
Identifying genes that meet criteria for reporting in NGSR and indication-based analyses
Our interdisciplinary group determined the set of rules for classifying each attribute. The following approach was used to ensure that genes associated with common genetic conditions that may present or be actionable during childhood were prioritized in the curation process. First, 430 genes defined as pediatric disease genes by Bell and colleagues5 were curated. Second, to identify genes associated with later onset or incomplete penetrance conditions for which action during childhood might prevent a devastating outcome, we searched for diseases known to our study team to be adult-onset or to have incomplete penetrance so they could be evaluated for validity and actionability. This search generated 113 genes, including 56 genes recommended by the American College of Medical Genetics and Genomics to be returned as incidental findings6 and additional genes associated with cardiomyopathy, cardiac conduction disease, breast and/or ovarian cancer, gastric cancer, nevoid basal cell carcinoma, melanoma, dyskeratosis congenita, pituitary adenoma, familial Alzheimer disease, Parkinson disease, amyotrophic lateral sclerosis, myofibrillar myopathy, spinal and bulbar muscular atrophy, oculopharyngeal muscular dystrophy, aceruloplasminemia, glycogen storage disorders, amyloidosis, and diabetes. Finally, to prioritize additional genes and accelerate indication-based analysis for ill newborns, lists of genes associated with 15 common newborn conditions encountered in Boston Children’s Hospital’s neonatal intensive care unit (hyperbilirubinemia, hypoglycemia, bowel dysfunction, hypothyroidism, hearing loss, respiratory disorder, inborn errors of metabolism, congenital heart disease, hypotonia, seizures, anemia/thrombocytopenia, thrombophilia, renal disease, skeletal dysplasia, and dermatologic disease) were created by assembling all genes reported in association with these presentations in the literature, through searching in OMIM (Online Mendelian Inheritance in Man), HGMD (the Human Genome Mutation Database), and PubMed. In addition, new genes were curated during the analysis of variants identified in BabySeq cases if the gene had not been previously curated. As of October 2016, curation of 1,514 gene–disease pairs has been completed, which includes 1,395 genes associated with one or more conditions (Supplementary Table S1 online). Curation of all genes associated with six common newborn disorders (hyperbilirubinemia, hypoglycemia, bowel dysfunction, hypothyroidism, hearing loss, and respiratory distress) has been completed. Curation of genes associated with the remaining nine common neonatal intensive care unit disorders is ongoing.
Based on criteria consistent with the ClinGen clinical validity framework, 34% of the gene–disease pairs had definitive, 33% had strong, 16% had moderate, and 16% had limited evidence for association ( Figure 2 ). Two genes, MYBPC3 and TMPO, had conflicting evidence for a causal role in dilated cardiomyopathy. The majority (94%) of the genes were associated with diseases presenting during childhood (<18 years), with 79% presenting at age ≤2 years, whereas only 6% presented during adulthood; however, it should be noted that the genes were selected primarily for pediatric presentation.

Summary of curated data for 1,514 gene–disease associations. The level of evidence that the gene is associated with disease, age of onset, and penetrance for all gene–disease pairs curated (top) and statistics for those with strong and definitive evidence (bottom) are demonstrated. Overall, 884 genes that have strong/definitive evidence to cause highly penetrant childhood-onset disease and 70 additional genes that are actionable in childhood met the BabySeq Project NGSR criteria, ~59.3% of which are typically inherited in an autosomal recessive manner and ~6.4% are inherited in an X-linked recessive manner.
Our understanding of penetrance is limited by the number of individuals screened for a particular gene or a variant in an unbiased manner and how well those individuals have been phenotyped. Therefore, our annotations of penetrance should be considered with this limitation. Based on the information available, approximately 73% of genes had high, 9% had moderate, and 1% had low penetrance. This classification was made with high confidence for 1,023 genes that had data from a large number of individuals (definitive or strong evidence) and with lower confidence for 246 genes with a smaller number of families (moderate evidence). We did not attempt to define penetrance for the 16% of curated genes that had limited or conflicting evidence.
Among the 1,023 genes with strong and definitive evidence for disease association, 97% were reported to present before age 18 years, with 81% presenting during infancy (≤2 years) ( Figure 2 ). Furthermore, 88% of these genes had high, 11% had moderate, and 1% had low penetrance.
Based on the results of our curation, we classified gene–disease pairs into three categories.
Category A: genes included in the NGSR with definitive or strong evidence to cause a highly penetrant childhood-onset disorder
Pathogenic variants in genes under this category have a high predictive value for a childhood-onset disorder and therefore meet our criteria to be returned in nGS. Return of such variants would provide more reliable information about risk for childhood-onset disease. Overall, 884 (58%) gene–disease pairs were in this category (Supplementary Table S1 online).
Category A includes four groups of genes (for a representative group of genes in category A, see Table 1 ). The first group is associated with diseases for which most affected individuals were symptomatic at birth or during the newborn period. For such diseases, GS may eliminate the need for extensive clinical tests and reduce diagnostic odysseys. The second group includes genes associated with disorders that presented soon after birth and had treatment opportunities available. Some of these disorders are detectable by tandem mass spectrometry or other assays and are currently tested by conventional NBS programs. The third group is associated with diseases that present during childhood and would benefit from early intervention but currently are not tested in conventional NBS programs, such as lysosomal storage diseases or immunodeficiencies that are not detectable by T-cell-receptor excision circle assays. Finally, the fourth group of genes present during childhood and have no effective treatment opportunities available at present, although knowledge at birth may still be beneficial to improve the health care and quality of life of the newborn and family. These benefits include timely supportive care, reducing the diagnostic odyssey when symptoms develop, preparation for the care of an ill child, and allowing family counseling and reproductive planning.
Category B: genes included in the NGSR based on actionability during childhood
This category consists of genes with moderate evidence or moderate penetrance for which professional guidelines or expert opinion determined that noninvasive interventions would be likely to improve the outcomes. Genes associated with diseases such as cardiomyopathies, cardiac conduction diseases, and certain cancer syndromes for which noninvasive screening during childhood may allow early detection were placed into this category.7,8,9,10,11 These genes may be included in the NGSR because the benefits of preventing a devastating outcome are likely to outweigh the uncertainty of disease risk. Two groups of genes were considered to fall into this category: (i) genes with moderate evidence and/or penetrance for which noninvasive intervention during childhood may prevent a devastating outcome and (ii) genes associated with diseases that typically present in adulthood but for which noninvasive intervention during childhood may significantly improve the clinical outcome (Supplementary Table S2 online). An example of a gene in this category is MYBPC3, which is associated with hypertrophic cardiomyopathy. MYBPC3 has definitive evidence for a causal role in hypertrophic cardiomyopathy, which may present during childhood.12,13,14,15,16,17,18,19 Although the penetrance is only moderate, knowing this risk at birth may allow routine surveillance by echocardiography and noninvasive interventions when needed, which would provide tremendous benefits for reducing the risk of sudden cardiac death.
Seventy genes were placed in category B. Overall, 954 of the curated 1,514 gene–disease associations (63%) were in categories A and B and therefore met criteria to be returned.
Category C: genes that did not meet criteria to be returned in the NGSR
This category consists of genes excluded from reporting in the NGSR either due to having insufficient (moderate/limited/no/conflicting) evidence to cause disease or having low/moderate penetrance, therefore having low predictive value, or due to being associated with adult-onset conditions for which there is no evidence that noninvasive intervention during childhood may improve outcome ( Table 2 ). These genes did not meet our reporting criteria for healthy newborns or as incidental findings for newborns with unrelated indications; however, they may be returned in an indication-based analysis if associated with the patient’s symptoms. If additional evidence supporting the gene’s role in disease or suggesting earlier disease onset becomes available in the future, then these genes may be reclassified for inclusion in the NGSR.
Use of the curated gene database in nGS analysis
To understand whether curating gene–disease association facilitates interpretation of genomic sequence data for nGS, we used our reference list in the NGSR analysis of the first 15 newborns sequenced in the BabySeq Project. Initial filtration identified variants with predicted loss of function or those reported in HGMD or ClinVar and having an allele frequency ≤3% in the general population. This approach resulted in 8 to 21 variants (median 14 variants) in genes associated with Mendelian disease per case, which were further assessed to determine whether they met criteria for NGSR inclusion. To ensure that variants in genes with recently published information and those that have not been previously curated were not missed, the curated gene list was not used in variant filtration. The gene list was used to accelerate variant analysis following the initial filtration described so that for each rare variant identified, only new information that would change the classification of the gene was reviewed if the gene had been curated previously. Otherwise, the gene was curated at the time of case analysis. In total, 201 variants were detected in 163 genes, with variants in 27 genes detected more than once. Of the 163 unique genes, 111 had already been curated and we checked whether any new information was available that would change their previous classifications. The remaining 52 genes were curated during the analysis of each case. Overall, 68/163 genes (42%) did not meet the criteria for return at the time of analysis ( Table 3 ). Based on results of gene curation, 83/201 variants (41%) were in genes not meeting NGSR criteria excluding them from further analysis.
Variants in 95/163 genes were further assessed for clinical significance and 18 were classified as pathogenic or likely pathogenic and returned (Supplementary Table S3 online). All of these variants were heterozygous and conferred carrier status for highly penetrant recessive childhood-onset disorders. No variants with strong evidence to suggest childhood-onset disease risk were identified. Eleven of 15 cases had at least one carrier status variant reported (six newborns had one, four had two, and one had four carrier status variants; four had none).
Assessment time for each rare variant ranged between 10 minutes and 4 hours, depending on the available literature. Excluding 41% of the median number of 14 variants/exome from further assessment saved several hours in the interpretation process.
Discussion
To accelerate interpretation and facilitate integration of GS into infant care, we curated 1,514 genes and classified them based on the appropriateness for return in nGS, using criteria established by our expert group. Our expectation is that future criteria for return of results in nGS will benefit from insights gathered in the BabySeq Project and similar studies.
The validity of gene–disease associations were classified using criteria based on the ClinGen clinical validity framework. It should be noted that the intent of the framework was to provide a provisional classification that is subsequently reviewed and finalized by disease experts; many of the validity assessments have not yet been approved by experts.
There are no guidelines for defining categories of age of onset or penetrance of genes. Therefore, we generated our own criteria to curate these attributes. Because the pathogenic mechanism and, thus, the expression of the phenotype may differ for each gene, we made our classifications at the gene level. The assertions regarding penetrance and age of onset were made with higher confidence for genes that had definitive or strong evidence due to the larger amount of data available for these genes. Our ability to evaluate these attributes was limited by the phenotype reports in the literature, which are undoubtedly biased toward diseased individuals due to the common use of families enriched with multiple relatives with the condition.
Among the 1,514 gene–disease associations curated, 32% did not have a strong or definitive level of evidence for a causal role in disease, suggesting that caution should be exercised in interpreting variants identified in these genes. Age of onset was younger than 18 years for 94% of curated genes. This large percentage is partly due to prioritization of genes associated with newborn presentations. Because we started with genes defined as disease-causing in the literature, none of the genes in our list were classified as having no evidence for a role in disease. As a result of this prioritization, the statistics of our curated list are likely to be different than those for a randomly selected list of genes. However, it is interesting that although genes associated with pediatric disorders were prioritized, only 63% met criteria to be returned in the NGSR. This result emphasizes the need for expert review of genes to determine appropriateness for reporting in nGS.
Although we established a strict set of criteria for the return of results, the decision regarding whether a gene met NGSR criteria was challenging for many genes. One such group of genes presented with a mixed phenotype that partially met reporting criteria. For example, there was moderate evidence that the FLNC gene is associated with myofibrillar myopathy, a disease including myopathy and cardiac arrhythmias. Although cardiac arrhythmias may be considered actionable during childhood, reporting pathogenic variants in this gene would disclose information regarding risk for adult-onset myopathy, which does not meet NGSR reporting criteria. Therefore, FLNC was considered as not meeting NGSR criteria. Another group of challenging genes had low penetrance; however, knowing that a person has pathogenic variants in them may be beneficial to avoid precipitating factors and may have a positive impact on the clinical outcome. An example is HMBS, which is an established gene for acute intermittent porphyria with low penetrance (approximately 10% are symptomatic). This gene does not meet reporting criteria due to low penetrance; however, learning about the risk for acute intermittent porphyria at birth may provide the opportunity to avoid precipitating factors and reduce the risk for acute attacks. As we learn more about the benefits and risks of returning such information in nGS, genes similar to FLNC and HMBS may be included in future nGS.
The majority of the 954 genes meeting NGSR criteria were associated with recessive conditions. This implies that if the carrier status for recessive disorders is reported in nGS, then it may have a significant impact on the number of individuals with reported variants. Indeed, carrier status variants were identified in the majority of the first 15 BabySeq cases. Returning carrier status in nGS may be helpful for the child and family members in future reproductive planning and may provide clinically significant information for some recessive disorders for which carrier individuals may have mild presentations. However, adequate genetic counseling is essential to ensure that results are well understood and that follow-up testing options are available for parents to estimate their reproductive risk. Although there is literature regarding the impact of returning carrier status information to children, these studies generally focus on adolescents with a family history of disease. There are currently no data regarding the implications of returning the carrier status of newborns. To understand the impact of returning carrier status information in nGS, we included this information in the NGSR in the BabySeq Project.
There is ongoing debate about genetic testing for adult-onset disorders in children and whether nondisclosure of adult-onset disease risk may do more harm to both the child and the family, especially if the disorder is actionable.20,21,22 One example is BRCA1 variants associated with breast cancer risk. It is true that there is currently no intervention in childhood known to impact the outcome of breast cancer. However, withholding this information may lead to the possibility of never receiving the result throughout their lifetime if later analysis is not pursued, thus losing the benefit of early intervention, which may be lifesaving. Furthermore, returning pathogenic variants in BRCA1 in a newborn would also disclose that one of the parents probably harbors the same variant. Early intervention based on this knowledge may have lifesaving consequences for the infant’s parent, which in turn could obviously impact the child’s quality of life. This subject will certainly continue to be discussed as the use of GS for children increases. Alternative approaches may include opt-in by the parents to receive adult-onset disease risk information for their newborn.
Although we periodically update our classifications for previously curated genes, because new information is published constantly, our reference list should not be considered final. Despite this limitation, the reference list was useful for facilitating results interpretation in the first 15 BabySeq cases and eliminated the need for manual variant assessment for 41% of detected rare variants in genes reported as Mendelian disease genes. This exclusion rate is expected to increase as additional genes are curated. Our curated list is available as Supplementary Table S1 online, and future updates will be made available online as a public resource.
Disclosure
O.C.-B. is an employee of the Mount Sinai Genetic Testing Laboratory, Icahn School of Medicine at Mount Sinai. T.W.Y. reports receiving consulting fees from and equity in Claritas Genomics outside of the submitted work. R.C.G. is supported by NIH grants and reports that he receives personal compensation for speaking or consulting from AIA, Helix, Illumina, Invitae, and Prudential. The other authors declare no conflict of interest.
References
Richards S, Aziz N, Bale S, et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424.
Duzkale H, Shen J, McLaughlin H, et al. A systematic approach to assessing the clinical significance of genetic variants. Clin Genet 2013;84:453–463.
McLaughlin HM, Ceyhan-Birsoy O, Christensen KD, et al.; MedSeq Project. A systematic approach to the reporting of medically relevant findings from whole genome sequencing. BMC Med Genet 2014;15:134.
Rehm HL, Bale SJ, Bayrak-Toydemir P, et al.; Working Group of the American College of Medical Genetics and Genomics Laboratory Quality Assurance Commitee. ACMG clinical laboratory standards for next-generation sequencing. Genet Med 2013;15:733–747.
Bell CJ, Dinwiddie DL, Miller NA, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med 2011;3:65ra4.
Green RC, Berg JS, Grody WW, et al.; American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013;15:565–574.
Gersh BJ, Maron BJ, Bonow RO, et al.; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Failure Society of America; Heart Rhythm Society; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011;124:2761–2796.
Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011;8:1308–1339.
Thakker RV, Newey PJ, Walls GV, et al.; Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012;97:2990–3011.
Calmettes C, Ponder BA, Fischer JA, Raue F. Early diagnosis of the multiple endocrine neoplasia type 2 syndrome: consensus statement. European Community Concerted Action: Medullary Thyroid Carcinoma. Eur J Clin Invest 1992;22:755–760.
American Gastroenterological Association. American Gastroenterological Association medical position statement: hereditary colorectal cancer and genetic testing. Gastroenterology 2001;121:195–197.
Morita H, Rehm HL, Menesses A, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med 2008;358:1899–1908.
Richard P, Charron P, Carrier L, et al.; EUROGENE Heart Failure Project. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003;107:2227–2232.
Van Driest SL, Vasile VC, Ommen SR, et al. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol 2004;44:1903–1910.
Olivotto I, Girolami F, Ackerman MJ, et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc 2008;83:630–638.
Alfares AA, Kelly MA, McDermott G, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med 2015;17:880–888.
Oliva-Sandoval MJ, Ruiz-Espejo F, Monserrat L, et al. Insights into genotype-phenotype correlation in hypertrophic cardiomyopathy. Findings from 18 Spanish families with a single mutation in MYBPC3. Heart 2010;96:1980–1984.
Watkins H, Conner D, Thierfelder L, et al. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet 1995;11:434–437.
Bonne G, Carrier L, Bercovici J, et al. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet 1995;11:438–440.
Ross LF, Ross LF, Saal HM, David KL, Anderson RR ; American Academy of Pediatrics; American College of Medical Genetics and Genomics. Technical report: Ethical and policy issues in genetic testing and screening of children. Genet Med 2013;15:234–245.
Botkin JR, Belmont JW, Berg JS, et al. Points to consider: ethical, legal, and psychosocial implications of genetic testing in children and adolescents. Am J Hum Genet 2015;97:6–21.
Wilfond BS, Fernandez CV, Green RC. Disclosing secondary findings from pediatric sequencing to families: considering the “benefit to families”. J Law Med Ethics 2015;43:552–558.
Emmerich J, Rosendaal FR, Cattaneo M, et al. Combined effect of factor V Leiden and prothrombin 20210A on the risk of venous thromboembolism–pooled analysis of 8 case-control studies including 2310 cases and 3204 controls. Study Group for Pooled-Analysis in Venous Thromboembolism. Thromb Haemost 2001;86:809–816.
Dahlbäck B, Zöller B, Hillarp A. Inherited resistance to activated protein C caused by presence of the FV:Q506 allele as a basis of venous thrombosis. Haemostasis 1996;26(suppl 4):301–314.
Bertina RM, Koeleman BP, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994;369:64–67.
Kjellberg U, van Rooijen M, Bremme K, Hellgren M. Factor V Leiden mutation and pregnancy-related complications. Am J Obstet Gynecol 2010;203:469.e1–469.e8.
Gohil R, Peck G, Sharma P. The genetics of venous thromboembolism. A meta-analysis involving approximately 120,000 cases and 180,000 controls. Thromb Haemost 2009;102:360–370.
Grody WW, Griffin JH, Taylor AK, Korf BR, Heit JA ; ACMG Factor V. Leiden Working Group. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med 2001;3:139–148.
Oji V, Seller N, Sandilands A, et al. Ichthyosis vulgaris: novel FLG mutations in the German population and high presence of CD1a+ cells in the epidermis of the atopic subgroup. Br J Dermatol 2009;160:771–781.
Nomura T, Akiyama M, Sandilands A, et al. Prevalent and rare mutations in the gene encoding filaggrin in Japanese patients with ichthyosis vulgaris and atopic dermatitis. J Invest Dermatol 2009;129:1302–1305.
Sandilands A, Terron-Kwiatkowski A, Hull PR, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet 2007;39:650–654.
Smith FJ, Irvine AD, Terron-Kwiatkowski A, et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet 2006;38:337–342.
Kamino K, Orr HT, Payami H, et al. Linkage and mutational analysis of familial Alzheimer disease kindreds for the APP gene region. Am J Hum Genet 1992;51:998–1014.
Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991;349:704–706.
Levy E, Carman MD, Fernandez-Madrid IJ, et al. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990;248:1124–1126.
Welcsh PL, King MC. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet 2001;10:705–713.
Tonin PN, Mes-Masson AM, Futreal PA, et al. Founder BRCA1 and BRCA2 mutations in French Canadian breast and ovarian cancer families. Am J Hum Genet 1998;63:1341–1351.
Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 1997;336:1401–1408.
Castilla LH, Couch FJ, Erdos MR, et al. Mutations in the BRCA1 gene in families with early-onset breast and ovarian cancer. Nat Genet 1994;8:387–391.
Acknowledgements
Research reported in this publication was supported by the National Institutes of Health under awards U19HD077671, R01HD075802, U01HG006500 and U41HG006834. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Consortia
Corresponding author
Supplementary information
Supplementary Information
(ZIP 491 kb)
Rights and permissions
About this article

Cite this article
Ceyhan-Birsoy, O., Machini, K., Lebo, M. et al. A curated gene list for reporting results of newborn genomic sequencing. Genet Med 19, 809–818 (2017). https://doi.org/10.1038/gim.2016.193
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.193
Keywords
This article is cited by
-
Abnormal biochemical indicators of neonatal inherited metabolic disease in carriers
Orphanet Journal of Rare Diseases (2024)
-
Genomic newborn screening for rare diseases
Nature Reviews Genetics (2023)
-
Genome screening, reporting, and genetic counseling for healthy populations
Human Genetics (2023)
-
Current attitudes and preconceptions on newborn genetic screening in the Chinese reproductive-aged population
Orphanet Journal of Rare Diseases (2022)
-
Application of next generation sequencing in the screening of monogenic diseases in China, 2021: a consensus among Chinese newborn screening experts
World Journal of Pediatrics (2022)
