
Abstract
Genetic testing for spinocerebellar ataxia is used in diagnosis of rare movement disorders. Such testing generally does not affect treatment, but confirmation of mutations in a known gene can confirm diagnosis and end an often years-long quest for the cause of distressing and disabling symptoms. Through interviews and a web forum hosted by the National Ataxia Foundation, patients and health professionals related their experiences with the effect of patents on access to genetic testing for spinocerebellar ataxia. In the United States, Athena Diagnostics holds either a patent or an exclusive license to a patent in the case of six spinocerebellar ataxia variants (spinocerebellar ataxia 1–3 and 6–8) and two other hereditary ataxias (Friedreich's Ataxia and Early Onset Ataxia). Athena has enforced its exclusive rights to spinocerebellar ataxia-related patents by sending notification letters to multiple laboratories offering genetic testing for inherited neurological conditions, including spinocerebellar ataxia. Roughly half of web forum respondents had decided not to get genetic tests. Price, coverage and reimbursement by insurers and health plans, and fear of genetic discrimination were the main reasons cited for deciding not to get tested. Price was cited as an access concern by the physicians, and as sole US provider, coverage and reimbursement depend on having payment agreements between Athena and payers. In cases in which payers do not reimburse, the patient is responsible for payment, although some patients can apply to the voluntary Athena Access and Patient Protection Plan offered by the company.
Similar content being viewed by others


Main
Views of patients and health professionals about the effect of patents on access to genetic testing for spinocerebellar ataxia (SCA) were sampled through a web forum. Questions about access to genetic testing were posted on the website of the National Ataxia Foundation (NAF) that agreed to host the web forum. The web forum was supplemented by interviews with three neurologists and a laboratory director. Both the web forum and interviews focused on perceptions of whether patents impede neurological patients from using diagnostic tests for the most common autosomal dominant forms of SCA.
SCA is not a single condition but is a group of progressive neurological genetic disorders with common symptoms and disparate genetic causes. Genetic testing plays a direct role in identifying the molecular defect in some cases.
Patents were generally not perceived as a major impediment in most cases. However, they were thought to have reduced access to the degree they increased the cost of testing borne directly by patients. Most SCA genetic testing in the United States is performed by Athena Diagnostics, which has licensed exclusive patent rights from various holders of SCA patents, mainly Universities and nonprofit research institutions. Some genes involved in SCA testing are not patented at all.
Price and concern about genetic discrimination both seem to reduce the use of genetic tests by patients. The decision not to get tested holds even for those who have progressed far into a diagnostic workup. SCA is a relatively rare syndrome and many genes are involved. The cost of testing can be as high as $7300, and only 6 of 14 surveyed patients who sought testing received reimbursement for testing from their insurance carriers. However, clinical testing requires Clinical Laboratory Improvement Amendments certification and testing for many genes, so patients are generally grateful that there is a test provider.
Athena offers a Patient Protection Plan (PPP) that caps out-of-pocket payments at 20% of the price for cases in which Athena directly bills the patient's insurer. However, this likely covers only a small minority (10–15%) of patients tested by Athena. Some patients who availed themselves of Athena's PPP appreciated the out-of-pocket payment caps. Under this program, Athena takes responsibility for seeking reimbursement from payers and insurers for the other 80%. Athena also has an “Athena Access” program for those who cannot afford the 20% copay that entails case-by-case review by Athena; analysis of Security and Exchange Commission (SEC) filings suggests that this covers relatively few patients. The 20% copay cap for patient outlays under the PPP is a standard option, although many patients seem not to know about it, and those in certain health programs are not eligible for it. Athena did not provide statistics on the percentage of patients covered by these two programs, so we are unable to estimate their actual effect. Although the program is discretionary and operated by Athena, independent data about how many people use it, which insurers and health programs are covered, and other details are not available because these details were not shared with us by the company.
We cannot estimate the population prevalence or magnitude of the effects because of the highly selected nature of our sample. However, the website respondents do indicate the two main reasons that patients with rare genetic conditions may not use genetic tests even when they would like to have the results of them: fear of discrimination and cost.
BACKGROUND
SCA is a rare subset of autosomal dominant cerebellar diseases characterized by loss of cerebellar cells. (SCA is also associated with other patterns of inheritance, both autosomal recessive and X-linked, but these are even more rare and less well characterized and therefore less relevant for commercial genetic testing.) Symptoms include ataxia, or irregular movement because of loss of neural control, and often additional symptoms attributable to loss of brainstem and spinal cord function.1,2 Ataxia is a common symptom found in conditions from chronic alcoholism to stroke. SCA accounts for <5% of the ataxic population, an estimated prevalence of 1–5:100,000.3–5 Although accounting for only 1 in 20 cases of ataxia, the autosomal dominant SCA syndromes are nonetheless much more common than the other genetic forms that are quite rare.
The severity and types of symptoms vary among the cerebellar ataxias. Diagnosis often takes years and may entail many visits to a succession of physicians and other health professionals, especially when it is a “proband case,” or the first known case in a family cluster. Once a first case is found in a family and confirmed as an inherited condition, genealogical tracing may identify other cases in the family. Currently, there are >30 identified variants of SCA (named SCA1–8 and 10–25, Table 1), a number that is constantly expanding. Each variant has the symptom of ataxia; however, the secondary symptoms can differ greatly. The symptoms associated with variants of SCA have significant overlap, so purely clinical symptom clusters rarely specify a particular mutation without DNA or protein testing. The vagueness with which each variant is described can make purely clinical diagnosis without genetic testing difficult, although there have been many attempts to design clinical scales using symptom correlations to hone in on a diagnosis (Fig. 1).6,7 There is no known cure for any subtype of SCA, and treatment often involves addressing individual symptoms as they appear.

Clinical scales used to determine SCA variants. Many neurologists have attempted to create neurological tests that can increase SCA diagnose accuracy before attempting genetic testing. Reprinted with permission from Mashke M, Oehlert G, Xie TD, et al. Clinical feature profile of spinocerebellar ataxia type 1–8 predicts genetically defined subtypes. Movement Disorders 2005;20:1405–1412. Copyright © 2005, John Wiley & Sons, Inc.
Before 1982, symptoms that now distinguish the various subtypes of SCA were diagnosed as olivopontine cerebellar atrophy, a classification that encompassed a host of cerebellar diseases.8 Later, a clinical classification was proposed for a subset of these diseases based on the mode of inheritance and secondary neurological symptoms not associated with the cerebellum.9 These classifications, known as the autosomal dominant cerebellar ataxias (ADCAs), were subdivided into classes I, II, and III. This diagnostic refinement allowed neurologists to distinguish ataxias according to their accompanying symptoms, among those being: peripheral nerve damage and dementia (ADCA I), macular and retinal degeneration (ADCA II), and an especially severe late-onset pure form of ataxia (ADCA III).
Eventually, as genetic research was incorporated into the classification along with symptoms, these conditions were divided further into >30 conditions now known as SCA.6,9,10 ADCA I was supplanted by SCA1–3, ADCA II was replaced with SCA7, and ADCA III was divided into SCA4–6 and 11. The classification has now become molecular. It is now based on mutations in genes that encode proteins affecting nerve function. Some mutations are associated with changes in proteins that conduct charged particles through cell membranes (“channel” proteins), receptors, or other surface proteins on nerve cells. Despite the increased specificity in diagnosis, treatment has changed little. Progress in understanding the molecular basis of SCA has not yet translated to better treatment. The primary benefits of SCA genetic testing for a patient are precision of prognosis, some reassurance from an accurate characterization of the molecular defect, an ability to diagnose those who are affected or presymptomatic among relatives, and the ability to test for a known mutation during preimplantation diagnosis or prenatal diagnosis in the progeny of an affected person. We focus here on the six most common forms, SCA1–3 and SCA 6–8.
Symptoms and pathology of SCA subtypes
SCA1 has pan-cerebellar symptoms that typically begin in a patient's fourth decade, with an average duration of 15 years (Table 1). Cerebellar symptoms include an atrophy of Purkinje cells, loss of afferent projections into the cerebellar cortex, and atrophy of dentatorubral pathways. Noncerebellar symptoms include signs associated with damage to the dorsal columns and cranial nerve nuclei. SCA1 maps to chromosome 6p23 in the ataxin-1 gene and is a CAG trinucleotide repeat disorder (i.e., caused by expanded number of those three base pairs inserted as repeats into the DNA of the gene). SCA1 is estimated to represent 5.6% of autosomal dominant SCA ataxias.3
In addition to cerebellar symptoms found with olivopontocerebellar atrophy, SCA2 is associated with marked loss or slowing of saccadic eye movements, dementia, and other peripheral neuropathy. Patients with SCA2 can show their first symptoms from the age of 2 to 65 years, a wide range in age of onset, with an average duration of 10–15 years from onset to death.11 SCA2 locus maps to the ataxin-2 gene in chromosome 12q24.1 and is a CAG trinucleotide repeat disorder. SCA2 is one of the most common of the ADCAs accounting for an estimated 15.2% of SCA occurrences.3
SCA3, also known as Machado-Joseph disease, is characterized by cerebellar signs including degeneration of the dentate and vestibular nuclei, with no degeneration of the cerebellar cortex. Additional symptoms include extrapyramidal, motor cranial nerve, anterior horn, posterior root ganglion, and spinopontine symptoms. SCA3 symptoms begin in the fourth decade with a typical duration of 10–15 years. SCA3 maps to chromosome 14q24.3-q32.2 in the ataxin-3 gene and is also a CAG trinucleotide repeat disorder. First described as Machado-Joseph disease, SCA3 accounts for 84% of autosomal dominant ataxias in ethnic Portuguese and 50% in ethnic German populations. In the United States, SCA3 accounts for ∼21% of dominant ataxias. The severity of symptoms correlates with the number of CAG repeats.12
SCA6 is an autosomal dominant ataxia that has a variable presentation, classified as three separate syndromes. The SCA6 syndromes are episodic ataxia, cerebellar ataxia plus brainstem or long tract degeneration, or pure cerebellar ataxia. Additional symptoms include a coarse gaze-evoked nystagmus, downbeat nystagmus on lateral gaze, and poor suppression of eye movement by vision.13 SCA6 is a slowly progressing late-onset version of SCA that begins in the fifth or sixth decade with a typical duration of >25 years. The SCA6 mutation has been identified as a CAG expansion located at 19p13, a subunit of the voltage-gated calcium channel locus CACNL1A4. SCA6 accounts for ∼15% of autosomal dominant ataxias in the United States.
In addition to cerebellar symptoms and ataxia, SCA7 is associated with retinopathy or blindness. Beginning in the third or fourth decade, SCA7 is a slowly progressive ataxia, lasting for an average of 20 years. Mapped to 3p21.1-p12, which encodes ataxin-7, SCA7 is a CAG repeat disorder as well. SCA7 accounts for roughly 5% of dominantly inherited ataxias in the United States. It is sometimes associated with genetic anticipation (earlier onset in successive generations, usually indicative of expansions of trinucleotide repeats) and severe childhood onset.
SCA8 is a less severe form of SCA, with symptoms including hyperreflexia, decreased sense of vibration, ataxic dysarthria (lack of control of joints), impaired smooth-pursuit eye movement, horizontal nystagmus, and atrophy of the cerebellar vermis and hemispheres. SCA8 begins in the fourth decade and is not associated with shortened lifespan. SCA8 has been mapped to 13q21 that encodes ataxin-8 and is a CAG/CTG repeat disorder. SCA8 accounts for 2–5% of autosomal dominant ataxias in the United States.
Diagnosing SCA
When first diagnosing or treating an ataxic patient, one of the first lines of evidence is family history. Similar to many diseases with known genetic causes, a family history that reveals multiple family members afflicted with similar clinical conditions can indicate that diagnosis of SCA should be considered.
Although an ataxic patient whose family history includes a genetically confirmed diagnosis of an SCA subtype is an ideal candidate for genetic testing, such cases are unusual. Most family histories contain no results from genetic testing because it is a relatively new technology. If ataxic symptoms exist in a family record, a previous diagnosis is likely to reflect a classification given to the disease at the time of diagnosis. Despite a dated classification, these histories are still valuable for diagnosing hereditary ataxias (Fig. 2).

Diagnostic tree for SCA. The diagnostic tree for SCA relies on many different tools. The most powerful tool remains to be family history, because this can quickly bring a patient to genetic testing. Because most ataxias are sporadic (not due to known inheritable factors), genetic testing does not occur early in the tree. Reprint with permission from Zumrova A. Problems and possibilities in the differential diagnosis of syndrome spinocerebellar ataxia. Neuro Endocrinol Lett 2005;26:98–108.
Family history is unavailable in many cases. A patient may be adopted, where heredity is impossible to trace through standard pedigree tracing. A patient may come from a family that has had little exposure to modern medicine and record keeping. Because of the late onset of some hereditary ataxias, not all families have had life expectancies long enough for symptoms to be observed.9 Many die without a definitive diagnosis, and stigma associated with uncontrollable movement can lead to cases being “hidden” from family discussion and result in incomplete pedigrees.
An initial neurological consultation is also intended to resolve whether the ataxia is acquired (e.g., related to alcohol use, infection, or other known syndromes) or sporadic. Acquired ataxia refers to cases with no known genetic component. These can be highly variable from case to case.9,10 Common causes of acquired ataxia include chronic alcoholism, stroke, multiple sclerosis, vitamin deficiency, and metabolic deficiencies.14–16 In some cases, identifying the source of an acquired ataxia can lead to a relief or even reversal of symptoms.
Sporadic ataxia is a diagnosis given to a subset of patients who have ataxia with no known hereditary or acquired components. This residual classification is broken into either pure cerebellar or “cerebellar plus,” depending on whether there are symptoms in addition to ataxia.6,10,17 Regardless of whether the ataxia seems to be hereditary, sporadic, or acquired on an initial evaluation, an ataxic patient will generally undergo a complete neurological evaluation and an magnetic resonance imaging scan.
After one or several magnetic resonance imaging scans, the neurologist may observe cerebellar atrophy or loss of cerebellar tissue. In addition to atrophy, the neurologist may observe signs and symptoms of a progressive loss of function in systems associated with the cerebellum. Symptoms can include gait disruption, nystagmus, vertigo, or general lack of coordination. Secondary noncerebellar symptoms including impaired cognition, memory, and vision can also point to SCA.
INTELLECTUAL PROPERTY LANDSCAPE—ATHENA DIAGNOSTICS
Many genetic tests for SCA are available only from Athena Diagnostics, including the most commonly used SCA genetic tests. There are currently 15 variants of SCA for which genetic testing is available. Athena Diagnostics holds the patent or has exclusive license to 12 patents that identify six SCA variants (SCA1–3 and 6–8) and two other hereditary ataxias (Friedreich's Ataxia and Early Onset Ataxia) included in their Complete Ataxia Panel (Table 2). These variants are the most commonly occurring, accounting for roughly 60–80% of known SCA cases, depending on the patient's country of origin.18–20 Athena was also granted a nonexclusive license by Baylor Medical College for US6855497, which covers methods for detecting SCA-10 (T. Rakow, Sr. Licensing Associate, Baylor Licensing Group, Baylor College of Medicine, personal communication, 2008). Athena also offers testing for SCA5, 13, 14, and 17.
Of the 12 patents listed by Athena, half of them are licensed from the University of Minnesota. Three others are from academic institutions (two through Research Foundation, one from Baylor) and only one is assigned to Athena itself. It thus seems that at least 9 of the 12 (75%) are licensed from academic institutions and one arose from in-house R&D at Athena.
Athena diagnostics has enforced its exclusive licenses and is widely assumed to be the sole laboratory for the above tests.21,22 The legal department of Athena has sent notification letters to some laboratories performing SCA genetic tests for which Athena has exclusive patent rights (Fig. 3). In another instance, the Diagnostic Molecular Pathology Laboratory at the University of California Los Angeles stopped offering testing for SCA 2 years ago, after receiving a letter from Athena Diagnostics. According to Dr. Wayne Grody, Director of the Laboratory, the terms of the sublicense offered by Athena Diagnostics were not economically viable for the laboratory (W. Grody, University of California Los Angeles, personal communication, 2008). Attempts to negotiate terms of a sublicense have not been successful to date. It is unclear to what extent cessation of testing at University of California Los Angeles has affected patient access to SCA testing. Dr. Grody indicated that samples are now sent to Athena Diagnostics for clinical testing. Several other laboratories are also listed on GeneTests.org for adult SCA diagnoses. Comprehensive Genetics Services offers a complete panel of SCA tests (Table 3), but did not respond to questions about patents or licensing in phone interviews. We became aware that Boston University reached a settlement with Athena Diagnostics regarding testing for SCA and several other conditions and no longer offers SCA testing (A. Milunsky, Director, Center for Human Genetics, Boston University, personal communication, 2008).

Example of a notification letter. Athena Diagnostics has protected their intellectual property rights using letters similar to the above.
Athena Diagnostics does not offer prenatal or preimplantation genetic diagnosis. Several laboratories listed on Gene-Tests.org perform these tests. We did not verify or otherwise pursue questions about prenatal or preimplantation genetic testing for SCA.
SCA genetic tests can be performed individually for as little as $400, for the least expensive single-locus test or as much as $2,335 for full-sequence analysis of the most expensive full-sequence gene test. (On February 10, 2010, Athena Diagnostics wrote to the Secretary's Advisory Committee on Genetics, Health, and Society, “SCA genetic tests can be performed individually for as little as $400, for the least expensive single-locus test, or as much as $1,170 for full-sequence analysis of the largest, full-sequence gene test” [T. Fenton, Thermo Fisher Scientific, which owns Athena Diagnostics, personal communication, 2010].) The lower cost tests are for known mutations in the second or subsequent members of a family, once a proband case in that family is characterized. Athena also offers the complete ataxia panel, a compilation of 18 tests that cover the most commonly identified SCA mutations for the price of $7300. This cost includes PCR tests and tests requiring sequencing (Table 3 and 4). The Athena price for the five most common SCAs (1, 2, 3, 6, and 7) is $2300 (if ordered individually, which is the only option). A university genetics laboratory can reportedly perform the same five SCA tests for $750 (or $1500 if ordered individually; personal communication with a laboratory directory who requested anonymity).
Most of the testing revenue of Athena Diagnostics comes from direct billing to hospital and commercial laboratories that send samples to Athena for patients seen or tested within their healthcare systems. In a public filing with the SEC, Athena indicated that 85% of their revenue comes from this source.23 This is widely known in the clinical laboratory industry as the most profitable type of billing arrangement: the sending facility is obligated to pay the full contractual price of testing directly to Athena regardless of insurance coverage or patient ability to pay; the sending facility then bills the patient and/or the patient's third-party payer for the cost of testing. Although reimbursement for this type of testing is often low or absent, the financial burden of poorly reimbursed testing is thus transferred from Athena to the sending healthcare facility and to the patient.
Athena has a formal policy that limits out-of-pocket expenditures for some patients for whom Athena directly bills the insurer. The PPP of Athena charges the patient 20% of the test fee (the usual copayment for most insurance programs) up front. After completing the test, if insurance covers the cost of the test, Athena will reimburse the patient for any payment their insurance makes 80% of the total bill. Such patients must have private insurance. If the patient's insurance does not cover the genetic test, Athena will limit the patient's liability to the 20% already paid. This plan requires preapproval by Athena. There is an additional plan for low-income families, Athena Access. This is for those who may find the 20% copay prohibitive. Athena, on receiving a request for a test, will attempt to contact the patient by mail and phone three times to enroll them into the PPP. Athena did not provide specific numbers regarding enrollment percentages into the PPP. It is also not clear whether this program includes persons whose only coverage is Medicare or Medicaid. However, the information in the SEC filing cited above would indicate that this program could apply to no more than 15% of the sources of revenue for testing at Athena in 2001. The PPP will not provide relief to a patient being billed by a healthcare facility for testing sent to Athena under the common direct billing arrangement accounting for 85% of the revenues of Athena. There is no way from public sources to estimate how much of the remaining 15% of revenue might have been reduced by the PPP or Athena Access programs.
Athena also has a repeat customer program that can reduce costs borne by patients. If a patient has a genetic test performed by Athena and receives a positive result, subsequent family members who request the same test have a greatly reduced price. However, the impact of this program is probably small because of the low rate of positivity for SCA testing (only 6% for patients without a known family history of SCA).24
The benefit of Athena licensing SCA patents from several different academic institutions and combining them with their own patent is that this enables a single laboratory to test for many variants. It also protects the investment of the company in CLIA certification, laboratory proficiency testing, a sales force to educate neurologists about the tests, and staff to manage the complex coverage and reimbursement policies of a dizzying array of disparate payers, insurers, and health plans. The syndromes are relatively rare, and it is possible that this full range of tests would not be available without the patent incentive.
The counter argument is that Athena has consolidated IP into an effective temporary monopoly for genetic testing of the SCA syndromes. It has been vigilant in enforcing its patent rights, and this has led several laboratories to avoid SCA testing, who otherwise might have offered a service alternative. As with all patented inventions, this reduces price competition, means all samples must be sent to an external laboratory, limits alternatives for verification of test results, and reduces the incentive to introduce cheaper and faster tests, because the current technology is protected by patents. For example, this could reduce incentives to develop a chip-based or microarray-bead or sequence-based test using alternative technologies, because the patents apply to any technology for assessing patented mutations or diagnostic methods that entail sequencing or sampling a patented sequence. Sole provider status also means that Athena effectively becomes the only testing service for mutations never yet detected (or patented), because the nature of the mutation is not known when a sample is sent. This means a single private firm becomes the repository for data needed to determine whether a discovered DNA change is actually a disease-causing pathogenic mutation or a benign polymorphism, information that is critically important to clinical interpretation. Yet, Athena does not seem to publish or report such data, leaving reporting to the disparate groups sending samples to their central laboratory.
Many of these tests were developed through federally funded research and licensed to Athena. The ultimate payer is often the federal government (through Medicare, Medicaid, Federal Employee Health Benefit Plan, Tricare, Veterans Health Administration, military health systems, Indian Health Service, etc.). The patents arising from federally funded research are subject to Bayh-Dole government use rights. Those government use rights are clearly not being interpreted to cover even payments channeled through the same federal Department of Health and Human Services that houses the National Institutes of Health that funded the research (DHHS payers include Medicare and federal components of Medicaid, the Indian Health Service and any genetic services covered by the Health Resources and Services Administration). We do not know whether there is a price reduction for National Institutes of Health-funded clinical trials and clinical research, because none of the respondents in the web forum specifically noted participation in such trials and the physicians interviewed did not mention this as well.
PHYSICIAN UTILIZATION AND ACCESS (INTERVIEWS WITH NEUROLOGISTS)
To get clinicians' perspectives on access to genetic testing, we interviewed three neurologists, with varying degrees of expertise with SCA, and a laboratory director. The neurologists we interviewed are Dr. Octavio de Marchena from the Neurology Associates of Lynchburg, Lynchburg, VA; Dr. James Burke from Duke University Hospital, Durham, NC; and Dr. Thomas Bird from the University of Washington, Seattle, WA. The laboratory director requested that his/her name be withheld and that interview is protected by a certificate of confidentiality (as are others, except when they were explicitly “on the record”), under the institutional review board-approved interview protocol we followed.
Dr. de Marchena is a general neurologist at a regional hospital. He treats all types of neurological patients and refers cases of ataxia to a subspecialist as needed. Confirmed cases of hereditary ataxia treated by Dr. de Marchena are rare, but he has had patients for whom he has established a positive diagnosis of SCA using genetic testing.
Dr. James Burke is a neurologist who specializes in neurodegenerative diseases at Duke, a private hospital and clinical outpatient service that is part of a major regional academic medical center. Dr. Burke does not solely treat patients with movement disorders, but does have ataxic patients referred to him from both inside and outside Duke. Cases of SCA are also rare for Dr. Burke, although he orders an estimated 5–10 genetic tests for SCA per year.
Dr. Thomas Bird is a researcher and clinician at the University of Washington and VA Puget Sound Health Care System, an academic health center with a long and distinguished history of medical genetics. He is an international expert on SCA and his research includes the genetics of neurodegenerative diseases. His patients are often referred to him from all over the country, and many come with the expectation that they will have genetic tests performed as a part of their visit. Compared with the other neurologists, Dr. Bird's patients are more often prescreened as candidates for genetic testing. Many patients referred to him have been seeking a specific diagnosis for some time. Many become involved in research studying trinucleotide repeat neurological diseases. Dr. Bird uses genetic tests much more often than most other neurologists. He estimated that in a given year he prescribes genetic testing for 35–45 ataxic patients, most patients receiving testing for multiple variants. When itemized by SCA variant, the number of individual tests that he orders comes to >200 per year. He is also a consultant to Athena.
We asked all the three neurologists to describe their use of genetic testing for SCA and the medical factors most responsible for prescribing tests. We asked them about how they interact with Athena Diagnostics. In addition, we asked them to describe their interactions with insurance companies and how insurance and health plan factors affect their use of genetic testing for SCA. Finally, we asked whether and how reducing the price of testing to $100 might change their use of genetic testing for SCA.
Clinical guidelines and utility of genetic testing
Achieving a diagnosis of SCA is more a complex process than a formal algorithm. The primary reasons for this are that ataxia is a common symptom associated with many disorders and because there are numerous forms of SCA. It takes substantial evidence from multiple methodologies (family history, brain imaging, and blood tests) over several visits, often documented in medical records from different providers, before a neurologist considers a genetic test for SCA. The only time this is not the case is if the family history contains a specific SCA diagnosis in another family member. (In these cases, although a test will come earlier, there is still no guarantee of a positive result.)
Considering that genetic testing provides the sole confirming diagnosis of SCA, we probed further the rationale for delaying genetic tests until after significant clinical evaluation. The primary reason is low likelihood that genetic testing will be informative in symptomatic ataxia that is not fully characterized (by ruling out alcohol use, stroke, or multiple sclerosis, for example). Even the most common form of SCA in US populations (SCA3) is likely to test negative >99% of the time in a patient displaying ataxia without a family history.3 If a neurologist can follow-up disease progression long enough, he or she can discern whether it follows any of the identified classifications of SCA, increasing likelihood of a positive genetic test. Clinical heterogeneity even among patients afflicted with the same variant of SCA can make it difficult for a neurologist to identify the SCA genes that should be tested.
Cost and cost effectiveness enter into decisions about genetic testing for SCA, but not in a simple way. Although positive results on genetic tests for SCA subtypes provide definitive diagnosis for ataxia in a patient, the interpretation of a negative result is much less well defined, and yet, negative results are common, even among well-screened patients. Many patients with clinical ataxia do not have a mutation in any of the genes known to be associated with SCA. In such cases, the diagnosis will be a clinical, descriptive, or anatomic one, such as olivopontine cerebellar atrophy.
Cost effectiveness of genetic testing
The primary issue associated with genetic testing for SCA is the low rate of positive results. The 30 identified SCAs only account for an estimated 5% of all diagnosed ataxias.3–5 Genetic tests are available for 12 of the genetic subtypes, representing an estimated 65–80% of SCA cases (Table 1). Edlefsen et al.24 compared the cost effectiveness of genetic tests. In this study, 162 patients were given a total of 282 neurogenetic tests. The patients were referred for genetic testing by a neurologist based on their family history and symptoms. The tests included mutations associated with chorea, neuropathy, muscle weakness, and ataxia. Of all, 30.2% of patients received a positive result on a genetic test or panel of tests corresponding to their symptoms. When only looking at probands, patients for whom there is no known family history, the positive rate goes down to 21.5%. For tests related to SCA (SCA1–3, 6–8, 10, 12, and 17), the total positive rate for patients was 11% (2/18), and only 5.9% (1/18) for probands without known family history of other SCA cases. This “hit” rate for SCA was the lowest of all genetic tests surveyed.
One way to assess testing cost is to estimate the cost per positive test result. The genetic test for Huntington disease costs $300. With a positive result in 71.2% of tests, the cost per positive is $440. For the Huntington disease test, the symptoms, family history of chorea, and need to test only a single locus makes selection of a genetic test straightforward. For the SCA tests, the cost of the test itself varied from $225 for a single-locus test (for a known mutation in a second or subsequent family member) to $2335 for a test requiring sequencing.24 Twenty-seven genetic tests for SCA, ordered for 18 patients, were either for single variants (17 of 27; SCA3, 7, 8, 10, 12, 14, and 17) or as a panel (10 of 27; SCA1–3, & 6–7). With a positive result rate of 11%, the cost per positive test for SCA was $7620, the most expensive cost per positive test studied. Edlefsen et al.24 noted that this cost would increase to >$50,000 if all tests were sent to Athena Diagnostics.
Adoption by clinical providers
When asked how they obtained SCA genetic testing for a patient, the neurologists said that they simply check a box on a requisition form. A blood sample goes with the form to the pathology department of their institution. From there, the in-house laboratory either ships the sample to Athena Diagnostics or performs tests, depending on the test. All the three neurologists stated that the testing from Athena was generally consistent and reliable. The neurologists all stated their personal preference was that the laboratory of their own institution would perform these tests, especially those that are PCR based and do not require sequencing.
Neurologists judged that the price of the test was sometimes problematic, mainly because insurance would not always reimburse all costs, and patients were not always able to cover the remaining costs. They considered cost as a factor but focused primarily on the clinical value of genetic testing. The patient might decide to forego testing, but while these neurologists considered costs, they saw their main task as explaining the clinical value of a genetic test, and left final determinations about whether a test was worth the cost to their patients.
To probe price sensitivity, the neurologists were asked a question about whether a decrease in test pricing to $100 would increase test prescriptions. All the three neurologists reiterated their stance that the limitations of current SCA genetic testing panels place genetic testing for this condition low on the diagnostic tree and late in the process, so such testing is not common and therefore not a major cost driver for diagnosis overall. They indicated that lowering the price to $100 would have little effect on their prescribing pattern. Dr. de Marchena stated that “any neurologist would call for testing when the symptoms and family history call for it, regardless of price.” Price is not the main factor in deciding whether to test, although it is a factor they consider. Dr. Bird noted that any neurologist must take into account “what value is this to a patient and his family, just giving the test without thought will not benefit them.” However, on further reflection, Dr. Bird believed that he would order more tests if the cost was substantially lower.
Lowering the price of testing would not affect the informational value of the test, as neurologists focus on “benefit to the patient,” and indeed it may be appropriate for patients to decide for themselves the value of the genetic test, because there are no clinical treatments that follow from specific genetic diagnosis. The neurologists order the test to provide clinically relevant information; the patients must then decide the personal value of that information to them, compared with their out of pocket costs and any other costs (needing to deal with applying for the Patient Protection program, Athena Access, etc.). The benefits of testing are mainly that the diagnostic work-up can end with a definitive result, a genetic diagnosis enables more precise prognosis, and it enables risk evaluation and a much more efficient diagnostic strategy for others related to that person.
The article by Edelfsen et al. notes that testing for ataxia is among the most expensive areas for genetic testing and that costs would be even higher if patents were enforced rigorously. They conclude: “For example, the cost per positive result for ataxia testing would increase nearly 7-fold, to >$50,000, if all tests had been obtained from the laboratory (NB: clearly referring to Athena Diagnostics) that claims exclusive patent rights for many of these tests. This increase reflects both higher per test cost and test packaging that encourages the ordering of larger panels of tests. Thus, policymakers should be aware that many of the costs per positive result found in this study may be greatly increased in the future because of intellectual property restrictions.”24
Adoption by third-party payers
Although price does not seem to have a strong effect on the number of tests ordered by neurologists, the contribution of insurance may influence whether patients go ahead and get genetic testing. When insurance does cover costs for Complete Ataxia Panel of Athena, generally leaving an estimated 20%, or $1500 copay, almost all patients who were not personally opposed to the test would take it. If insurance refused coverage, and patients were required to pay Athena the full price of $7300 for the Complete Ataxia Panel, both Dr. Bird and Dr. Burke reported that patients were likely to pay for the test less than half the time. Additionally, Dr. Burke stated that “for individual tests for a specific SCA variant, insurance often covered the request because the evidence of its utility was much greater.” Dr Bird noted that $7300 self-pay is prohibitive for most patients.
The neurologists all concurred about the inconsistency in insurance companies deciding to cover a genetic test. The uncertainty surrounding insurance decisions sometimes led to their postponing genetic testing while awaiting insurer preapproval and often having to write time-consuming letters of justification.
PATIENT PERSPECTIVES
We solicited direct patient input through a web forum. The mission of the NAF (www.ataxia.org) is to improve the lives of those suffering from ataxia by offering information, support, and resources. The NAF maintains a bulletin board forum with >700 users, many of whom have an ataxia or are family members of someone who does. The forum supplies information about where to go for diagnosis and how to cope with the effects of disease. With the cooperation of the NAF, we established a discussion thread on this forum and asked users to discuss their personal stories regarding genetic testing for ataxia.
We began the discussion with a list of questions about genetic testing, prices, the involvement of insurance and health plans, and how the results of the tests affected the patients. The questions are listed below, followed by a discussion of the responses. The participants were fully aware that they were participating in a public forum. The entire discussion through 17 October 2007 is given in the Appendix.
One purpose of the web survey and online forum was to convey information about patient perspectives to the SACGHS task force.
The response was impressive, with 30 responses and 450 views of the website over several weeks. The responses indicated that patients were passionate about the issue of access to genetic testing, and their comments provided insight into complexities of genetic testing that complemented the issues raised by the neurologists, the laboratory director, insurance companies, and policy makers.
Who Responded?
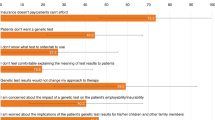
Among the 16 forum users who responded to our questionnaire, there were two major groups: those diagnosed with a variant of SCA and others (many of them were caregivers or family members). This is almost surely a highly biased, relatively well-informed, and therefore unrepresentative sample. For our purposes of getting knowledgeable and informed patient perspectives, however, it was an effective sample. Of the 16 respondents, 11 had an SCA genetic test performed. Of the five responders without a test, one abstained for fear of genetic discrimination, one would have a test performed soon, and the other three were not covered by their insurance and could not pay for the testing themselves. Of the 11 responders that took the test, six were covered by their insurance carrier and the others paid out of pocket. The users who had a diagnosis achieved it through genetic testing by Athena Diagnostics. Those without an SCA diagnosis either took the test and had negative or inconclusive results, or did not take a genetic test.
Responses regarding insurance deciding on coverage sounded a consistent theme: inconsistency in coverage and reimbursement decisions by payers (insurers and health plans).
Dancingpoodle wrote, “The insurance company said they wouldn't cover the genetic testing since there was no family history and the cost was so high.”
Jonab wrote, “I have called my insurer to see if I am covered, and they have told me that I am covered, if it's ‘medically necessary.'”
Rrose wrote, “My insurance company did not cover the cost of the test. The cost to me was $2500. They told me at the time it was because Athena was not one of their preferred providers. I was required to pay the entire amount upfront, directly to Athena Labs.”
Should genetic testing have been prescribed earlier?
Users were generally well informed about the various diseases presenting as ataxia and the limitations surrounding current diagnostic methodology. Most users had ataxia or lived with someone who did. They understood that more than two dozen variants of SCA had been identified, 12 of which could be genetically tested. They also understood and agreed that these tests should not be prescribed as a screening test for ataxia because a substantial clinical threshold needed to be crossed before a genetic test was warranted. When asked if their neurologist should have prescribed a genetic test earlier, three patients responded that while they would prefer to have the diagnosis made clear earlier, their neurologist ordered genetic testing at the appropriate time. Two patients stated that they went through multiple neurologists to get genetic testing ordered although both came up negative for a known SCA variant.
How has genetic testing for SCA affected you?
Having an undiagnosed progressive neurological condition is frightening and disheartening; yet, the users on the forum seemed patient. Diagnosis was an important uncertainty in the lives of many forum participants. Many had been seeking a diagnosis of their symptoms for years. Many did not have a diagnosis for their ataxia and other symptoms, despite having undergone extensive diagnostic evaluation, including genetic testing. Despite this, patients encouraged one another regularly to continue the quest for precise diagnosis. In their point of view, a positive or negative result on a diagnostic test helped and also advanced medical practice for future patients. They fully understood the lack of cure or prevention for any type of SCA and viewed their participation as essential to change this.
For participants who received a positive result on a genetic test for SCA, many described complex emotions. A positive result can give knowledge about the disease and its prognosis, but there is no cure.
Marjorienye wrote, “First, when I was diagnosed and then more so as my symptoms have worsened, I've felt more and more helpless to fight what is happening to me. I like to at least be somewhat knowledgeable about the disease that's wringing the freedom out of my life, since there's very little I can do to fight it. There's no surgery I can have, no experimental drugs, and rehabilitative techniques will only slow things down, not cure me. Sometimes it seems like knowledge is all I can depend on.”
Many of the positive results came from users who had a relative with a specific form of SCA already diagnosed through genetic testing. The result was particularly difficult if the patient was asymptomatic (meaning that the test was presymptomatic) or if they had children of their own.25,26
Rrose stated, “Having a definite diagnosis is helpful in some ways, as I tend to focus my research, but troubling in other ways with respect to my children. They know I have the same thing as their grandmother, but the whole question of when to tell them they can be tested is very difficult. How do you tell three young men 20, 18, and 16 with no symptoms, to have testing done that might change the course of their life decisions? I'm not sure I have the answer to that.”
The benefits of a positive result included certainty of diagnosis, clearer prognosis, and information relevant to family planning decisions. The variable severity of SCA among subtypes meant that knowing which type a patient has could have a significant impact on almost every aspect of their life. For example, a patient with SCA6 can expect slow deterioration with relatively mild secondary symptoms.6 The SCA6 patient may be able to continue working a job not requiring much physical exertion. However, a diagnosis of SCA3 carries a much worse prognosis. A patient can expect to lose mobility in 5–10 years and face rapid progression of secondary symptoms that often leave the patient unable to work. For a younger or asymptomatic patient, a diagnosis of SCA3 may change long-term life planning. Dr. Burke reflected that some patients reevaluate their lives based on the expected years of functionality. Some patients ask him, “Why should I go to college if I know that in 20 years I'll be in a wheelchair?”
Do you feel you have access to genetic testing taking into consideration financial constraints?
A majority of the responses from the forum stated that even with genetic tests costing as much as $7300, genetic testing was accessible. In some cases, patients only required a single genetic test for a specific SCA variant. These tests, when performed by Athena, cost the patients from $88 to $440 depending on insurance reimbursement and PPP enrollment. Four of five patients who had a single test performed felt that the price was reasonable even if the out-of-pocket expense was the full $480 cost of the test, that is, without insurance coverage or price reduction through the PPP.
For a patient who did not have a positive diagnosis in their family history, the Complete Ataxia Panel, with its $7300 price tag, might be the prescribed diagnostic test. If insurance covered the test or the patient successfully enrolls in the PPP, the patient responsibility was $1500. There were relatively few complaints about the price on the forum. However, some comments implied that $1500 would inflict hardship on their family, especially considering the likely negative result.
Dancingpoodle wrote, “I suppose if I felt the test would help cure me if I knew what I had, I would take out a loan to have it done, but since there are no cures at the moment, I don't see a reason for putting that financial burden on my family.”
Some paid for a complete ataxia panel without any contribution from insurance or the PPP. Such patients included some who got testing from sources other than Athena. Another group of people seemed to be eligible for PPP of Athena but were unaware of the price reduction available. They assumed that denial by insurance was the end of the story, and both the patient and neurologist were unaware of the possibility of negotiating with Athena. In these cases, the price of $7300 reduced testing, with five of nine patients who were rejected by their insurer deciding to forego it. This indicates that both patients and Athena could benefit from greater coverage and reimbursement, and more knowledge about payment assistance and forgiveness programs.
The perceived risk of genetic discrimination is one unfortunate feature of SCA genetic testing. The survey was done before the Genetic Information Nondiscrimination Act (GINA) passed in May 2008 (and it did not begin to take effect until mid-2009, in any event).
Some who sought testing and might have been eligible for PPP of Athena abstained from using their insurance because they did not want their insurance company to know that they were being tested for SCA. These patients voluntarily decided to pay for testing out-of-pocket and therefore did not qualify for the PPP and often did not qualify for financial hardship reductions through Athena Access either. Although respondents on the forum surely did not reflect the general public, but highly selected individuals, it seemed the number foregoing genetic testing might be a significant number among those who would have found clinical value in the information available from the test. Of patients surveyed who had genetic testing and had insurance coverage for the testing, four of seven patients chose not to notify their insurer (to avoid genetic discrimination for themselves or others in their family). In such cases, the $7300 price did seem to result in some people choosing not to get tested. Whether genetic discrimination actually occurred, as no one reported an actual case of it, perceptions of the risk clearly did lead to decisions not to seek genetic testing. Several respondents were hopeful that GINA could alleviate health insurance and employment discrimination based on genetic testing if it became law, as it did in May 2008.
Following are the experiences of two respondents describing their hesitation about getting genetic testing. One patient (poolgirl) had the testing covered by insurance and then wished she had not done so because of the possible repercussions if she had tested positive for an SCA variant.
Poolgirl wrote, “I had the genetic tests done at a very vulnerable point in my work-up and thankfully they were negative. Given the implications a positive test could have on my children, had I been thinking clearly, I would not have done the tests or would have considered paying for them myself to avoid having them on record. I will not do any tests that become available in the future unless one of my children specifically requests it be done to help guide them if/when they are thinking of having children and if so, I would probably do it off the record. I have no problems with my medical insurance but my personal interaction with my disability company has made me very cynical about trusting any insurance carrier to do the right thing.”
Another story shows how one patient did not seek insurance reimbursement for her genetic test out of fear that her family would be labeled as a result. The loss of insurance for future generations was a major concern.
We note that in this case, health insurance discrimination from a Medicare carrier is unlikely because Medicare is an entitlement and does not entail medical underwriting. This response may reflect an incomplete understanding of Medicare. However, it is possible that SunnyKay was worried about how a Medicare reimbursement record might affect insurance status of a younger member of the family not in Medicare. However, it is unlikely that a specific Medicare genetic testing reimbursement decision would affect underwriting, independent of other information potentially in the medical record and available for any medical underwriting involving another family member. This perception of high risk of genetic discrimination is therefore probably not an accurate assessment or real risk, but it also shows that perception of risk can heavily influence the choice to get a genetic test.
SunnyKay wrote, “My mother requested that Athena not bill Medicare because she wanted to keep the results private for numerous reasons. That is why a payment plan was arranged instead. My mother not only did not want the health insurance company/Medicare to know about possible SCA results, she did not want to apply for a handicapped license plate with a diagnosis of some form of ataxia either. This is in addition to other things she did to keep anyone from the government or any other unnecessary place from finding out about my Dad's medical history, which of course becomes our and her children's family medical history for life.”
CONCLUSIONS
-
Both neurologists and patients stressed that inconsistency in coverage and reimbursement by payers was a common problem that has real consequences for patients and their families by reducing access to genetic tests for SCA.
-
All three neurologists interviewed agreed that a prescription for SCA genetic testing was primarily based on best medical practice, and the clinical value of information, not price. Despite their belief that prices of Athena are higher than if SCA were available in their home institution's laboratory, they did not describe any reduction in recommending genetic testing, and it was unclear whether lowering the price of testing to $100 would increase the number of tests they ordered. However, they noted that price did affect their patients, who must decide if the value of the genetic testing information is worth the cost, and so high price reduces utilization (and if this information is deemed clinically useful, then also access).
-
The neurologists emphasized the care they must take to ensure that they never give a diagnosis of SCA lightly, either positive or negative. An incorrect diagnosis can have devastating consequences. From the neurologists' perspective, one advantage of sending samples to Athena for testing is that the liability risks associated with the tests themselves are then borne by Athena. However, this transfer of liability would apply equally to testing sent to any clinical laboratory facility and is not unique to Athena.
-
When applicable, PPP of Athena can reduce the financial outlay out-of-pocket, and Athena Access is also available for case-by-case review of hardship. This feature was cited in the patient forum as valuable and may help explain the relative dearth of complaints about pricing of Athena. The actual use of this program is unknown, but statistics from 2001 stock offering of Athena suggest that 85% of revenues derive hospital and other facility billing, rather than direct patient or insurance payments. Not all patients know about PPP of Athena that can reduce patient out-of-pocket outlays to 20% of test price. Some who do know about it choose not to avail themselves of it for fear of losing health insurance. However, the reported financial data of Athena suggest that this program is likely applicable to only a small part of the revenue stream of Athena (10–15%) and probably a roughly comparable fraction of patients. It is also possible that some patients may not complain about test costs because most of the unreimbursed cost of testing is absorbed by the institutions that are billed directly for testing by Athena.
-
From the patient's perspective, the main benefit of in-house testing would be any decrease in cost and direct connection between the clinician ordering the tests and the laboratory performing them. Unlike some other case studies, we do not have lists of prices for test providers other than Athena. One published study estimated that the cost of ataxia testing at an institution would increase nearly 7-fold if all patented tests were referred to Athena rather than being performed by the institution's laboratory.24 Another estimate of testing price for the five most common SCA mutations was $1500 for a university laboratory compared with $2300 for Athena.
-
Use of genetic testing is reduced by fear of genetic discrimination by insurers, health plans, and employers that leads some not to seek third-party reimbursement for genetic tests. This was a factor for roughly half of those who had insurance coverage, and clearly led to some choices to not get genetic testing despite valuing the clinical information that would result from the tests.
-
We cannot discern in most cases whether this fear of genetic discrimination is warranted or merely perceived. Given the long search for a diagnosis in most cases, and thus the accumulation of medical records that would in theory be available to insurers and employers, it could be that the risk attributable to genetic testing specifically is lower than perceived— not necessarily because the risk is not there, but because risks of exclusion from health insurance, disability insurance, employment, or long-term care insurance are present even without the specific action of taking a genetic test. Seeking reimbursement does trigger payer scrutiny and so the risk of genetic discrimination that some respondents attributed to genetic testing could still be real. Patients expressed a desire for GINA to become law, echoing the calls from the Genetic Alliance and other groups. However, passage of the act affects only employment and health insurance, but not other forms of insurance for disability, life, and long-term care.
-
Not all patients know about the PPP of Athena that can reduce patient out-of-pocket outlays to 20% of test price. Some who do know about it choose not to avail themselves of it for fear of losing health insurance.
-
The laboratory director believed that exclusive licensing of patented tests created significant barriers to patient access. He believed that academic institutions exclusively licensing patents to single-source providers were short sighted and did not take into account that a university can achieve an equivalent royalty stream without giving exclusive control of their patents to a single company. He asserted that academic institutions should not accept an exclusive license bid for technologies that can readily enter the market. He believed that this was especially true for patents on many diagnostic tests in which the scientific advance may simply be a new combination of nucleotides used as a primer for a previously unidentified gene.
REFERENCES
Schols L, Bauer P, Schmidt T, Schulte T, Riess O . Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 2004; 3: 291–304.
Taroni F, DiDonato S . Pathways to motor incoordination: the inherited ataxias. Nat Rev Neurosci 2004; 5: 641–655.
Moseley ML, Benzow KA, Schut LJ, et al. Incidence of dominant spinocerebellar and Friedreich triplet repeats among 361 ataxia families. Neurology 1998; 51: 1666–1671.
Mori M, Adachi Y, Kusumi M, Nakashima K . A genetic epidemiological study of spinocerebellar ataxias in Tottori prefecture, Japan. Neuroepidemiology 2001; 20: 144–149.
van de Warrenburg B, Sinke RJ, Verschuuren-Bemelmans CC, et al. Spinocerebellar ataxias in the Netherlands: prevalence and age at onset variance analysis. Neurology 2002; 58: 702–708.
Maschke M, Oehlert G, Xie T, et al. Clinical feature profile of spinocerebellar ataxia type 1–8 predicts genetically defined subtypes. Mov Disord 2005; 20: 1405–1412.
Schmitz-Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006; 66: 1717–1720.
Konigsmark BW, Weiner LP . The olivopontocerebellar atrophies: a review. Medicine (Baltimore) 1970; 49: 227–241.
Harding A . The clinical features and classification of the late onset autosomal dominant cerebellar ataxias. A study of 11 families, including descendants of the ‘the Drew family of Walworth.’. Brain 1982; 105( Pt 1): 1–28.
Manto MU . The wide spectrum of spinocerebellar ataxias (SCAs). Cerebellum 2005; 4: 2–6.
Auburger G, Diaz GO, Capote RF, et al. Autosomal dominant ataxia: genetic evidence for locus heterogeneity from a Cuban founder-effect population. Am J Hum Genet 1990; 46: 1163–1177.
Onodera O, Idezuka J, Igarashi S, et al. Progressive atrophy of cerebellum and brainstem as a function of age and the size of the expanded CAG repeats in the MJD1 gene in Machado-Joseph disease. Ann Neurol 1998; 43: 288–296.
Gomez CM, Thompson RM, Gammack JT, et al. Spinocerebellar ataxia type 6: gaze-evoked and vertical nystagmus, Purkinje cell degeneration, and variable age of onset. Ann Neurol 1997; 42: 933–950.
Gordon N . Cerebellar ataxia and gluten sensitivity: a rare but possible cause of ataxia, even in childhood. Dev Med Child Neurol 2000; 42: 283–286.
Hadjivassiliou M, Grunewald R, Sharrack B, et al. Gluten ataxia in perspective: epidemiology, genetic susceptibility and clinical characteristics. Brain 2003; 126( Pt 3): 685–691.
Zumrova A . Problems and possibilities in the differential diagnosis of syndrome spinocerebellar ataxia. Neuro Endocrinol Lett 2005; 26: 98–108.
Koeppen AH . The pathogenesis of spinocerebellar ataxia. Cerebellum 2005; 4: 62–73.
Tang B, Liu C, Shen L . Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch Neurol 2000; 57: 540–544.
Lee WY, Jin DK, Oh MR, et al. Frequency analysis and clinical characterization of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Korean patients. Arch Neurol 2003; 60: 858–863.
Bauer PO, Zumrova A, Matoska V, et al. Absence of spinocerebellar ataxia type 3/Machado-Joseph disease within ataxic patients in the Czech population. Eur J Neurol 2005; 12: 851–857.
Schissel A, Merz JF, Cho MK . Survey confirms fears about licensing of genetic tests. Nature 1999; 402: 118
Cho MK, Illangasekare S, Weaver MA, Leonard DG, Merz JF . Effects of patents and licenses on provision of clinical genetic testing services. J Mol Diagn 2003; 5: 3–8.
Athena Diagnostics S-1 (Registration statement, SEC accession no.0000950130–01-504992). 2001; Available at: www.secinfo.com/dsvrt.4fD5f.f.htm. Accessed January 14, 2009.
Edlefsen KL, Tait JF, Wener MH, Astion M . Utilization and diagnostic yield of neurogenetic testing at a tertiary care facility. Clin Chem 2007; 53: 1016–1022.
Villiers Cd, Weskamp K, Bryer A . The sword of Damocles: the psychosocial impact of familial spinocerebellar ataxia in South Africa. Am J Med Genet 1997; 74: 270–274.
Smith CO, Lipe HP, Bird TD Impact of presymptomatic genetic testing for hereditary ataxia and neuromuscular disorders. Arch Neurol 2004; 61: 875–880.
Acknowledgements
The case study authors have no consultancies, stock ownership, grants, or equity interests that would create financial conflicts of interest. The Center for Genome Ethics, Law and Policy accepts no industry funding. Dr. Robert Cook-Deegan is listed on the British Medical Journal roster of physicians who have pledged to remain independent of industry funding (http://www.tseed.com/pdfs/bmj.pdf); more details about how the case studies were done are noted in a 29 July 2009 letter to the Secretary's Advisory Committee on Genetics, Health, and Society (http://www.genome.duke.edu/centers/gelp/documents/SACGHSResponsetopubliccomments.pdf).
All interviews were conducted under Duke University Institutional Review Board-approved protocol 1277 and usually conducted by phone and recorded. Researchers obtained informed consent from subjects. These interviews are covered by a federal certificate of confidentiality.
This case study was carried out under Grant P50 003391, co-funded by the National Human Genome Research Institute and US Department of Energy, and supplemented by funding from The Duke Endowment. Ashton Powell is now also part of the CEER research Grant P50 HG004488 at the University of North Carolina, Chapel Hill. This case study was reviewed by Michael Henry, Athena Diagnostics; Jonathan Tait, University of Washington; Octavio de Marchena, Neurology Associates, Lynchburg, VA; James Burke, Duke University; Michael Hopkins, Sussex University; and Thomas Bird, University of Washington.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest. See Acknowledgments for details.
APPENDIX














Rights and permissions
About this article
Cite this article
Powell, A., Chandrasekharan, S. & Cook-Deegan, R. Spinocerebellar ataxia: Patient and health professional perspectives on whether and how patents affect access to clinical genetic testing. Genet Med 12 (Suppl 1), S83–S110 (2010). https://doi.org/10.1097/GIM.0b013e3181d67e44
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181d67e44
Keywords
This article is cited by
-
Global trends on fears and concerns of genetic discrimination: a systematic literature review
Journal of Human Genetics (2016)
-
DNA patents and diagnostics: not a pretty picture
Nature Biotechnology (2010)
