
Abstract
Purpose: Phenylketonuria (PKU) is an autosomal recessive disorder caused by mutations in the phenylalanine hydroxylase (PAH) gene. There have been more than 400 mutations identified in the PAH gene leading to variable degrees of deficiency in PAH activity, and consequently a wide spectrum of clinical severity. A pilot study was undertaken to examine the response to 6-R-l-erythro-5,6,7,8-tetrahydrobiopterin (BH4) in patients with atypical and classical PKU.
Methods: PAH gene mutation analysis was performed using denaturing gradient gel electrophoresis and gene sequencing. Patients with classical, atypical, or mild PKU were orally given BH4 10 mg/kg. Blood phenylalanine and tyrosine levels were determined using tandem MS/MS at 0 hours, 4 hours, 8 hours, and 24 hours intervals.
Results: Thirty-six patients were given a single oral dose of 10 mg/kg of BH4. Twenty one patients (58.33%) responded with a decrease in blood phenylalanine level. Of the patients that responded, 12 were classical, 7 atypical, and 2 mild. The mean decline in blood phenylalanine at 24 hours was > 30% of baseline. There were 15 patients who did not respond to the BH4 challenge, 14 of those had classical and one had atypical PKU. Mapping the mutations that responded to BH4 on the PAH enzyme showed that mutations were in the catalytic, regulatory, oligomerization, and BH4 binding domains. Five patients responding to BH4 had mutations not previously identified.
Conclusion: The data presented suggest higher than anticipated number of PKU mutations respond to BH4, and such mutations are on all the domains of PAH.
Similar content being viewed by others


Main
Phenylketonuria (PKU) is a disorder caused by deficiency of the enzyme phenylalanine hydroxylase (PAH).1 The gene for PAH has been cloned and over 400 mutations have been identified.2,3 The findings of Bickel et al.,4 that a phenylalanine (Phe)-restricted diet can ameliorate the effects of high blood Phe on cognitive function, has resulted in newborn screening programs for PKU.5 Early results of treatment were very encouraging. However, treatment has to be continued “for life,” otherwise high blood Phe levels lead to functional deficits.6 Present treatment goals for PKU include maintaining blood Phe levels between 120 and 360 μmol/L. It has been difficult to comply with this treatment.7–17 In order to maintain optimal mental functioning in patients with PKU, a search for better methods of treatment has been on going. These methods include enzyme therapy, competition with transport of Phe to the brain, and potentially gene therapy.18–22
A new development in the treatment of PKU was reported by Kure et al.23 in four patients with mild PKU who responded to an oral load of the cofactor for PAH, 6-R-1-erythro-5,6,7,8-tetrahydrobiopterin (BH4). These patients had normal levels and production of BH4. After this report, other cases from Europe have been reported.24–30 These reports emphasize the potential response to BH4 of patients with atypical or mild PKU. Recently Matalon et al.31 presented favorable response to BH4 in a small number of patients with classical PKU.
This pilot study was undertaken in two clinics in the United States to find out the extent of response to BH4 among patients with PKU. The study included patients with classical, atypical, and mild PKU.
MATERIALS AND METHODS
The study was performed at two sites, the Children's Hospital of Los Angeles and University of Texas Medical Branch at Galveston. Informed consent was obtained from all participants or their parents. The study included 16 males and 20 females with PKU. There were 26 subjects with “classical” PKU, based on several quantitative determination of blood Phe levels > 1200 μmol/L after positive newborn screening for PKU, and also according to daily Phe intake of 150 to 250 mg. In the 8 patients with “atypical” PKU, blood Phe was 360 to 1200μmol/L, and daily Phe tolerance was 350 to 450 mg. In the two patients with “mild” PKU, blood Phe was 120 to 360 μmol/L. The mean age of the subjects was 16.6 years with a range from 6 months to 43 years. Twenty-seven of the 36 patients were taking low Phe diet, with various degrees of success in attaining blood Phe levels between 120 and 360 μmol/L. Only seven patients had blood Phe levels in the treatment range of 120 to 360μmol/L, whereas the other 20 patients had blood Phe levels above 360μmol/L. Six patients were not following a low Phe diet: one with classical PKU, four atypical, and one mild.
All of the patients had a normal urine pterin profile and blood dihydropteridine reductase (DHPR) activity. Urine pterin profile and blood DHPR were determined by Neogen (Bridgeville, PA). Mutation analysis on all patients was determined by Dr. Guttler using Denaturing Gradient Gel Electrophoresis (DGGE) of the coding sequence exons 1–12; and by sequencing of the exons with the mutations.32,33
Tetrahydrobiopterin, 6R-BH4, was obtained from Schircks Laboratories, Jona, Switzerland. Baseline blood Phe and tyrosine were drawn after an overnight fast and then the subjects were given BH4 10 mg/kg orally. Data from a single patient given 20 mg/kg BH4 were contributed by Dr. C. Scriver. Patients were instructed to continue the same dietary practice during the day after oral BH4 challenge. Blood Phe and tyrosine were taken at 4, 8, and 24 hours after an overnight fast. Blood analyses were determined using tandem MS/MS (Neogen, Bridgeville, PA). Analysis of variance (ANOVA) was used for statistical analysis.
Expression analysis
Escherichia coli expression and activity assessment experiments were performed on mutant AH protein expressed both with and without GroES and GroEL chaperonin coexpression (pGroESL) by the method followed earlier.34
For mammalian expression, wild-type and mutant PAH proteins were expressed using transient mammalian expression in COS cells and using an in vitro transcription-translation system (TnT-T7). Transient expression analysis in COS cells were performed using PAH cDNA cloned in the rRC/CMV expression vector (Invitrogen, CA) as followed earlier.35
RESULTS
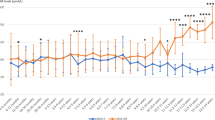
Twenty one subjects (58.33%) responded with a decline in blood Phe compared to the baseline levels. The mean blood Phe level before BH4 was 720.23 μmol/L, and after 24 hours BH4 loading, the mean level dropped to 423.33 μmol/L as shown in Table 1 (P = 0.0006). The mean decline at 24 hours in blood Phe was 58.77%. The maximum decline in blood Phe was at 24 hours in all patients except one who had maximum decline at 8 hours after BH4. The genotype and individual blood Phe response are shown in Table 1. Mean blood tyrosine levels before and 24 hours after the BH4 load are also shown in Table 1. Whereas tyrosine increased in some patients, the overall increase was not statistically significant.
Blood Phe and tyrosine before and after BH4 load in patients that did not respond are shown in Table 1. Two individuals who had either the R261Q or Y414C allele (IVS12nt1g>a/ R261Q and Y414C/IVS12nt1g>a) failed to respond to BH4. These mutations have been reported to cause favorable response to BH4. Two patients had a rise in blood tyrosine (IVS12nt1g>a/E280K and IVS12nt1g>a/R261Q), but overall there was no statistical significance in tyrosine response to BH4. The two patients with the R261Q allele were challenged again with 20 mg/kg BH4 and showed less than a 10% drop in blood Phe level.
The mutations in patients with favorable response to BH4 in this study are shown in Figure 1 mapped onto the structure of a composite of the monomer model of PAH. Five of these mutations have not previously been reported to respond to BH4:F39L/ F55fdelT, R68S/ R408W, H170D/IVS1nt5g>a, E178G/IVS10nt-11g>a, and L308F/R261Q.

C-Alpha trace of a monomer of the composite model of phenylalanine hydroxylase.51 Regulatory domain is colored in orange (residues 19–142), the catalytic domain in gray (residues 143–410), and the tetramerization domain (residues 411–452) in blue. Regions responsible for binding the BH4 cofactor are shown in purple. BH4 cofactor is shown together with the iron at the active site. Side chain of the residues found to be BH4 responsive are shown mapped onto the structure.
E. coli expression of mutation A313T, Y414C, and I65T showed 45%, 50%, and 91% residual activity, respectively.
DISCUSSION
Treatment of PKU has been undergoing reevaluation since the mid seventies. There have been an increased number of reports of intellectual decline, and white matter changes, in patients who stopped the dietary restriction of Phe.8,14
Neuropsychological and neuroimaging improvement have been reported when low Phe diet was resumed. At the same period of time, the problem of maternal PKU was looming, and the practice of diet discontinuation was questioned. A strong move toward “diet for life” has emerged. This change in treatment policy has been difficult to translate to the reality of lower blood Phe levels, especially when subjects with PKU enter the early teens.6
In an attempt to improve dietary compliance, medical foods for the treatment of PKU underwent many changes to improve taste and acceptability. New low protein foods have been developed, to be used as part of the low Phe diet plan. Such changes are still continuing. The “magic” formula and foods that a patient with PKU will readily take “for life” have yet to be found. Therefore, different approaches have been explored. Such ideas include the use of an enzyme, phenylalanine ammonia lyase (PAL), that breaks down Phe in the gastrointestinal tract.18–20 Another approach is to prevent the influx of Phe to the brain by the addition of large neutral amino acids (LNAA) to the diet as suggested by Kaufman.21 The LNAA include Phe, leucine, isoleucine, valine, tryptophan, tyrosine, methionine, threonine, and histidine. Studies using tyrosine, tryptophan, or branched chain amino acids and other short term trials with LNAA have been reported with no alternative treatment to Phe-restricted formulas.36–44
Treatment of PKU with BH4 is a novel approach started with the report of Kure et al. in 1999.23 Subsequently reports of challenging patients with PKU with BH4 have dealt with atypical or mild PKU.23–29,45 Our report is the first study showing a large number of patients with classical as well as atypical PKU responding favorably to a BH4 challenge by a drop in their blood Phe levels.31 Although “classical PKU” has been used to denote levels of blood Phe above 1200 μmol/L, it has become important to determine the genotype of patients with PKU, so that severity of the metabolic defect can be better correlated with the mutation and PAH residual activity. Of 12 subjects considered “classic” PKU, three had blood Phe levels > 1200 μmol/L and responded to BH4. The other patients had lower blood Phe levels because they were on some dietary restriction. In young patients who were on diet with good control of blood Phe, the drop of blood Phe level after BH4 load may not be as dramatic, but still was discernible.
A trial with BH4 supplementation on six patients with PKU showed improved tolerance to dietary Phe, suggesting the possibility of diet relaxation.46 Results of treatment of 38 PKU patients with BH4 showed that all the responders had mild PKU, and the six patients with classical PKU did not respond to BH4.47
Although the phenotype-genotype correlation in PKU is rather complex, the genotype of PKU patients who respond to BH4 may be predictive, although individual response may be different. Blood Phe and tyrosine have diurnal variation, so blood drawing needs to be done at similar times.48
Most of the mutations tested in this study have been reported in earlier BH4 trials.31,49–50 However, five previously unreported mutations have been found to be BH4 responsive. The BH4-responsive mutations F39L and R68S are located in the regulatory domain of PAH,51 whereas mutations H170D, E178G, and L308F are in the catalytic domain.
Mutation H170D is a newly discovered PKU mutation that is found in one of the patients of the study, together with IVS 1nt5g→a. Histidine 170 is located on the surface of the catalytic domain, close to the tetramerization domain and the regulatory domain. Substitution into aspartic acid may disrupt a hydrogen bond to Arg241 at the start of beta strand Cβ1.49 Mutation L308F is another newly discovered mutation found to be BH4 responsive in a patient along with the R261Q mutation on the second allele. Leucine 308 is close to the tetramerization domain residues valine 412 and tyrosine 414, and the catalytic domain residues alanine 259 and glutamic acid 305. When leucine is substituted with phenylalanine (L308F), there is no space for the phenylalanine side chain to be inserted, as would be the case in L308F. Substitution into a phenylalanine from a smaller leucine could potentially push the tetramerization domain away from the catalytic domain, and interfere with dimer/tetramer formation. Based on the BH4-responsiveness both the H170D and the L308F mutations may prove to be mild mutations with close to wild-type residual activity, but the mutations remain to be studied biochemically.
Based on the emerging large number of PKU mutations that are BH4 responsive, there does seem to be a few basic points that govern the BH4 response. The combination of alleles found in the PKU patient’s genotype seems to initially determine whether or not the patient shows BH4 responsiveness. If one of the patient’s alleles contains a mild mutation that does not completely abolish activity but imposes changes in cofactor binding (i.e., a Km mutant), this may be a sign of BH4 responsiveness. If both alleles contain severe mutations, it is unlikely that the patient will be BH4 responsive. A second option to explain the BH4 responsiveness could be the idea of a chemical chaperone affecting the function and stability of enzymes as suggested by Scriver and Waters.52 The BH4 cofactor may function as a chemical chaperonin by rescuing mutant PAH enzyme from host cell proteases (increasing the protein half-life), and making the enzyme able to perform its enzymatic functions for a longer period of time before being degraded.53 We have crystallized PAH with mutation A313T, and superimposed it on the wild-type PAH. Minor differences were noted in the crystal structure of BH4 binding domain. It has been proposed that BH4 responsiveness resulting from BH4 induced stabilization of mutant PAH dimers.54 The prerequisite for BH4 to function as a chemical chaperonin there has to be residual activity in vivo, with enzymatic activity in expression system. A third option for BH4 response is that BH4 up-regulates PAH gene expression,31,55 perhaps similarly to what is seen in the related and homologous enzyme tyrosine hydroxylase.56
Because PAH is a tetramer, the association of different monomers into a tetramer may vary depending on the mutations found in PAH, thus leading to functional hemizygosity that further leads to variable phenotype and variable response to BH4. More research is needed to clarify which mutations lead to better response to BH4. There is a need for more information from a new emerging discipline of genomic interaction and the effect on the proteome and the metabolome. Because the experiments presented in this study were short-term and only given at one time, it is evident that longer trial periods and double blind placebo control studies are needed in order to evaluate the effectiveness of BH4 in the treatment of PKU. As to management of PKU, BH4 loading is encouraged and long-term monitoring of response is needed. It seems that treatment with BH4 for all PKU patients may emerge if indeed tolerance to dietary Phe increases and blood tyrosine levels increase. Success of such a treatment should introduce a dramatic change in the way we manage PKU.
References
Scriver CR, Kaufman K . Hyperphenylalaninemia. Phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW et al, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill, 2001: 1667–1724.
Phenylalanine hydroxylase locus knowledge base, Montreal: McGill University health center, 2003. Available at: http://www.pahdb.mcgill.ca. Accessed October 31, 2003.
Nowacki P, Byck S, Prevost L, Scriver CR . PAH Mutation Analysis Consortium Database: 1997 Prototype for relational locus-specific mutation databases. Nucleic Acids Res 1998; 26: 220–225.
Bickel H, Gerard J, Hickmans EM . Influence of Phenylalanine intake on phenylketonurics. Lancet 1953; 2: 812–813.
Guthrie R, Susi A . A simple phenylketonuria screening method for newborn infants. Pediatrics 1963; 32: 338.
NIH Consensus Report on Phenylketonuria. “Phenylketonuria: Screening and management of PKU“. U.S. Department of Health and Human Services, Public Health Services, National Institutes of Health, National Institute of Child Health and Human Services, 2001.
Azen CG, Koch R, Friedman EG, Berlow S, Colwell J, Kruase W . Intellectual development in 12-year-old children treated for Phenylketonuria. Am J Dis Child 1991; 145: 35–57.
Holtzman NA, Kronmal RA, van Doorninck W, Azen C, Koch R . Effect of a great loss of dietary control on intellectual performance and behavior of children with Phenylketonuria. N Engl J Med 1986; 314: 593–598.
Fisch R, Chang P, Weisberg S . Phenylketonuric patients decades after diet. J Inherit Metab Dis 1995; 18: 347–353.
Thompson AJ, Smith IL, Brenton D . Neurological deterioration in young adults with Phenylketonuria. Lancet 1990; 336: 602–605.
Ris MD, Williams SE, Hunt MM, Berry HK, Leslie N . Early-treated phenylketonuria: Adult neuropsychological outcome. J Pediatr 1994; 124: 388–392.
Griffiths P, Paterson L, Harvie A . Neuropsychological effects of subsequent exposure to phenylalanine in adolescents and young adults with early-treated phenylketonuria. J Intellec Dis Res 1995; 39: 365–372.
Pietz J, Dunckelmann R, Rupp A, Rating D, Meinck HM, Schmidt H . Neurological outcome in adult patients with early-treated phenylketonuria. Eur J Pediatr 1998; 157: 824–830.
Koch K, Azen CG, Friedman EG, Williamson ML . Preliminary report on the effects of diet discontinuation in PKU. J Pediatr 1982; 100: 870–875.
Michals K, Azen C, Koch R, Acosta P, Matalon R . Blood phenylalanine levels and intelligence of ten year old children with phenylketonuria in the national collaborative study. J Am Diet Assoc 1988; 88: 1226–1229.
Schmidt E, Rupp A, Burgard P, Pietz J, Weglage J, de Sonneville L . Sustained attention in adult phenylketonuria: The influence of the concurrent phenylalanine-blood-level. J Clin Exp Neuropsychol 1994; 16: 681–688.
Burgard P, Rey F, Rupp A, Abadie V, Rey J . Neuropsychologic functions of early treated patients with phenylketonuria, on and off diet: Results of a cross-national and cross-sectional study. Pediatr Res 1997; 41: 368–374.
Sarkissian CN, Shao Z, Blain F, Peevers R, Su H, Heft R . A different approach to treatment of Phenylketonuria: Phenylalanine degradation with recombinant Phenylalanine ammonia lyase. Proc Natl Acad Sci 1999; 96: 2339–2344.
Hoskins JA, Ambrus JL, Jack G, Peiris RJD, Starr DJT, Wade HE . Enzymatic Control of Phenylalanine Intake in PKU. Lancet 1980; 1: 392–394.
Ambrus CM, Anthone S, Horvath C, Kalghatgi K, Lele AS, Eapen G . Extracorporeal enzyme reactor for depletion of Phenylalanine in PKU. Ann Intern Med 1987; 106: 531–537.
Kaufman S . Phenylketonuria: Biochemical mechanisms. In: Agranoff BW, Aprison MH, editors. Advances in Neurochemistry. New York: Plenum Press, 1977: 1–132.
Matalon R, Surendran S, Michals-Matalon K, Quast M, Jinga W, Ezell E . Future role of large neutral amino acids in transport of phenylalanine into the brain. Pediatrics 2003; 122: 1570–1574.
Kure S, Hou DC, Ohura T, Iwamoto H, Suzuki S, Sugiyama N . Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency: A novel clinical entity. J Pediatr 1999; 135: 375–378.
Spaapen LJM, Bakker JA, Velter C, Loots W, Rubio ME, Forget PP . Tetrahydrobiopterin-responsive hyperphenylalaninemia (HPA) in dutch neonates. J Inherit Metab Dis 2000; 23 ( suppl 1): 45.
Trefz F, Blau N, Aulehla-Scholz C, Korall H, Frauendienst-Egger G . Treatment of mild phenylketonuria (PKU) by Tetrahydrobiopterin (BH4). J Inherit Metab Dis 2000; 23( suppl): 47.
Trefz F, Aulela-Scholz C, Blau N . Successful treatment of phenylketonuria with tetrahydrobiopterin. Eur J Pediatr 2001; 160: 315.
Linder M, Haas D, Zschocke J, Burgard P . Tetrahydrobiopterin responsiveness in phenylketonuria differs between patients with the same genotype. Mol Genet Metab 2001; 73: 104–106.
Lindner M, Steinfeld R, Burgard P, Schulze A, Mayatepek E, Zschocke J . Tetrahydrobiopterin sensitivity in German patients with mild phenylalanine hydroxylase deficiency. Hum Mutat 2003; 21: 400.
Blau N, Trefz F . Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency: possible regulation of gene expression in a patient with the homozygous L48S mutation. Mol Genet Metab 2002; 75: 186–187.
Weglage J, Grenzebach M, Teeffelen-Heithoff T, Marquardt R, Feldmann J, Denecke D . Tetrahydrobiopterin responsiveness in a large series of phenylketonuria patients. J Inherit Metab Dis 2002; 25: 321–322.
Matalon R, Koch R, Michals-Matalon K, Moseley K, Stevens R . Tetrahydrobiopterin-responsive phenylalanine hydroxylase mutations. J Inher Metab Dis 2002; 25( suppl): 23.
Guldberg P, Henriksen KF, Guttler F . Molecular analysis of phenylketonuria in Denmark: 99% of the mutations detected by denaturing gradient gel electrophoresis. Genomics 1993; 17: 141–146.
Guttler F, Azen C, Guldberg P, Romstad A, Hanley WB, Levy HL . Relationship among genotype, biochemical phenotype, and cognitive performance in females with phenylalanine hydroxylase deficiency: report from the Maternal Phenylketonuria Collaborative Study. Pediatrics 1999; 104: 258–262.
Gamez A, Perez B, Ugarte M, Desviat LR . Expression analysis of phenylketonuria mutations: Effect on folding and stability of the phenylalanine hydroxylase protein. J Biol Chem 2000; 275: 29737–29742.
Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, Northrop JP . Lipofectin: A highly efficient, lipid-mediated DNA transfection procedure. Proc Natl Acad Sci USA 1987; 84: 7413–7417.
Lou H, Guttler F, Lykkelund C, Bruhn P, Niederwieser A . A decreased vigilance and neurotransmitter synthesis after discontinuation of dietary treatment for phenylketonuria in adolescents. Eur J Pediatr 1985; 144: 17–20.
Pietz J, Landwehr R, Kutscha A, Schmidt H, de Sonneville L, Trefz FK . Effect of high-dose tyrosine supplementation on brain function in adults with phenylketonuria. J Pediatr 1995; 127: 936–943.
Oldendorf WH, Szabo J . Amino acid assignment to one of three blood-brain barrier amino acid carriers. Am J Physiol 1976; 230: 94–98.
Pardridge WM . Blood-brain barrier amino-acid transport: Clinical implications. In: Cockburn F, Gitzelmenn R, editors. Inborn errors of metabolism in humans. Lancester, England: MTP Press Limited, 1982: 87–99.
Berry HK, Brunner RL, Hunt MM, White PP . Valine, isoleucine, and leucine: A new treatment for phenylketonuria. Am J Dis Child 1990; 144: 539–543.
Berry HK, Bofinger MK, Hunt MM, Phillips PP, Guilfoile MB . Reduction of cerebrospinal fluid phenylalanine after oral administration of valine, isoleucine and leucine. Pediatr Res 1982; 16: 751–755.
Hommes FA . The role of the blood-brain barrier in the aetiology of permanent brain dysfunction in hyperphenylalaninaemia. J Inherit Metab Dis 1989; 12: 41–46.
Dotremont H, Francois B, Diels M, Gillis P . Nutritional value of essential amino acids in the treatment of adults with phenylketonuria. J Inherit Metab Dis 1995; 18: 127–130.
Pietz J, Kreis R, Schmidt H, Meydig-Lamadz UR, Rupp A, Bosch C . Phenylketonuria. Findings at MR imaging and localized in vivo H-1 spectroscopy of the brain in patients with early treatment. Radiology 1996; 201: 413–420.
Bernegger C, Blau N . High frequency of tetrahydrobiopterin-responsiveness among hyperphenylalaninemias: a study of 1919 patients observed from 1988 to 2002. Mol Genet Metab 2002; 77: 304–313.
Hennermann JB, Vetter B, Kulozik AE, Monch E . Partial and total tetrahydrobiopterin-responsiveness in classical and mild phenylketonuria (PKU). J Inherit Metab Dis 2002; 25( suppl 1): 21.
Muntau AC, Roschinger W, Habich M, Demmelmeir P, Hoffmann B, Sommerhoff CP . Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N Engl J Med 2002; 347: 2122–2132.
Guttler F, Olesen ES, Wamberg E . Inverse diurnal variations of serum phenylalanine and tyrosine in phenylketonuric children on low-phenylalanine diet. In: Allan JD, Holt KS, Ireland JT, Pollitt RJ, editors. Enzymopenic Anaemias, Lysosomes and other papers, E&S. Edinburgh, Ireland: Livingstone LTD, 1969: 149–159.
Erlandsen H, Stevens RC . A structural hypothesis for BH4 responsiveness in patients with mild forms of hyperphenylalaninaemia and phenylketonuria. J Inherit Metab Dis 2001; 24: 213–230.
Steinfeld R, Kohlschutter A, Zschocke J, Lindner M, Ullrich K, Lukacs Z . Tetrahydrobiopterin monotherapy for phenylketonuria patients with common mild mutations. Eur J Pediatr 2002; 161: 403–405.
Erlandsen H, Stevens RC . The structural basis of phenylketonuria. Mol Genet Metab 1999; 68: 103–125.
Scriver CR, Waters PJ . Monogenic Traits are not simple: Lessons from Phenylketonuria. Trends Genet 1999; 15: 267–272.
Gamez A, Perez B, Ugarte M, Desviat LR . Expression analysis of phenylketonuria mutations: Effect on folding and stability of the phenylalanine hydroxylase protein. J Biol Chem 2000; 275: 29737–29742.
Steinfeld R, Kohlschutter A, Ullrich K, Lukacs Z . A hypothesis on the biochemical mechanism of BH4-responsiveness in phenylalanine hydroxylase deficiency. Amino Acids 2003; 25: 63–68.
Hyland K, Munk-Martin TL . Tetrahydrobiopterin regulate tyrosine hydroxylase and phenylalanine hydroxylase gene expression in dominantly inherited GTP cyclohydrolase deficiency. J Inher Metab Dis 2001; 24( suppl 1): 30.
Sumi-Ichinose C, Urano F, Kuroda R, Ohye T, Kojima M, Tazawa M . Catecholamines and serotonin are differently regulated by tetrahydrobiopterin: A study from 6-pyrovoyltetrahydrobiopterin synthase knock-out mice. J Biol Chem 2001; 276: 41150–41160.
Acknowledgements
This work has been supported by NICHD Grant No. R03 HD 40898, The Danish Medical Council Grant No. 9902901, and the Children’s Hospital Los Angeles Research Institute. The authors would like to acknowledge the contribution of one patient added to this series by Dr. Charles Scriver and Dr. Robert Barnes. The authors deeply appreciate the many helpful suggestions made by Dr. Charles Scriver.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Matalon, R., Koch, R., Michals-Matalon, K. et al. Biopterin responsive phenylalanine hydroxylase deficiency. Genet Med 6, 27–32 (2004). https://doi.org/10.1097/01.GIM.0000108840.17922.A7
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/01.GIM.0000108840.17922.A7
Keywords
This article is cited by
-
Assessment of Functioning among Nigerians with Schizophrenia in a Tertiary Health Facility: Influence of Psychopathology, Socio-demographic and Treatment Factors
Journal of Psychosocial Rehabilitation and Mental Health (2015)
-
Tetrahydrobiopterin responsiveness after extended loading test of 12 Danish PKU patients with the Y414C mutation
Journal of Inherited Metabolic Disease (2010)
-
Blood phenylalanine concentrations in patients with PAH‐deficient hyperphenylalaninaemia off diet without and with three different single oral doses of tetrahydrobiopterin: Assessing responsiveness in a model of statistical process control
Journal of Inherited Metabolic Disease (2009)
-
The Missense p.S231F Phenylalanine Hydroxylase Gene Mutation Causes Complete Loss of Enzymatic Activity In Vitro
The Protein Journal (2009)
-
Effects and clinical significance of tetrahydrobiopterin supplementation in phenylalanine hydroxylase‐deficient hyperphenylalaninaemia
Journal of Inherited Metabolic Disease (2007)
