
Abstract
Purpose: Mucopolysaccharidosis type I (MPS I) is an autosomal recessive disorder resulting from a deficiency of the lysosomal glycosidase, α-l-iduronidase (IDUA). Patients with MPS I present with variable clinical manifestations ranging from severe to mild. To facilitate studies of phenotype-genotype correlation, the authors performed molecular studies to detect mutations in MPS I patients and characterize single nucleotide polymorphism (SNP) in the IDUA gene.
Methods: Twenty-two unrelated MPS I patients were subjects for mutation detection using reverse transcriptional polymerase chain reaction (RT-PCR) and genomic PCR sequencing. Polymorphism analyses were performed on controls by restriction enzyme assays of PCR amplicons flanking nine IDUA intragenic single nucleotide polymorphic alleles.
Results: Eleven different mutations including two common mutations (Q70X, W402X), five recurrent mutations (D315Y, P533R, R621X, R628X, S633L), and four novel mutations (R162I, G208D, 1352delG, 1952del25bp) were identified from MPS I patients. Multiple SNP alleles coexisting with the disease-causing mutations were detected. Allelic frequencies for nine SNP alleles including A8, A20, Q33H, L118, N181, A314, A361T, T388, and T410 were determined.
Conclusions: The results provide further evidence for the mutational heterogeneity among MPS I patients and point out possible common haplotype structures in the IDUA gene.
Similar content being viewed by others

Main
Mucopolysaccharidosis type I (MPS I, MIM 252800) is an autosomal recessive disorder caused by various lesions in the α-l-iduronidase (IDUA) gene. A wide range of clinical manifestations in MPS I patients have been recognized and three major clinical subtypes, Hurler syndrome (MPS IH, severe), Hurler-Scheie syndrome (MPS IH/S, intermediate), and Scheie syndrome (MPS IS, mild), have been defined. MPS IH represents the severe type with clinical features of organomegaly, heart disease, severe mental retardation, and death in childhood. In contrast, MPS IS exhibits mild somatic complications, normal intelligence, and a normal life span. MPS IH/S represents a phenotype of intermediate clinical severity. All three MPS I subtypes are a result of the enzymatic deficiency in the lysosomal glycosidase IDUA (EC3.2.1.76). The deficient IDUA causes a disruption of the sequential degradation of the glycosaminoglycans, dermatan sulfate and heparan sulfate, and results in lysosomal accumulation and excessive urinary excretion of partially degraded dermatan sulfate and heparan sulfate.1,2 Routine in vitro identification of deficient IDUA enzyme activity provides a definitive biochemical diagnosis for MPS I, but the residual enzyme activity cannot correlate with the clinical subtypes. The characterization of human IDUA cDNA sequence3 and genomic structure4 has made possible the identification of mutations underlying different MPS I subtypes.5 Several studies have revealed the mutational diversity and ethnic/geographic heterogeneity in MPS I patients. Two common mutations, W402X and Q70X, were found in more than 60% of Caucasian mutant alleles,6 mutations 704ins5 and R89Q accounted for 42% of Japanese alleles,7 and mutation P533R was identified in 92% of mutant alleles in Moroccan patients.8 In addition, inasmuch as multiple polymorphic alleles in the IDUA gene have been detected in MPS I patients and controls, it is reasonable to hypothesize that a SNP in combination with a disease-causing mutation could produce a modification in the MPS I phenotype or catalytic activity of the protein in controls.9 Recently, the correlation of IDUA activity with polymorphic haplotypes was tested in a Chinese population.10 Analyses by immunoquantitation and enzyme kinetics of IDUA in cultured fibroblasts from normal controls and MPS I patients indicate a high level of genetic heterogeneity in IDUA gene where mutant alleles present different effects in IDUA protein quantity, substrate affinity, and catalytic activity.11,12 Therefore, molecular characterization of the mutational spectrum and polymorphism distribution in the human IDUA gene is important for achieving reliable diagnosis of clinical subtypes, improving prognostic prediction, providing accurate carrier detection and informative genetic counseling, and developing better preventive and therapeutic approaches.
As an initial effort to provide mutation analysis for MPS I patients enzymatically diagnosed in our laboratory, an approach based on reverse transcriptional polymerase chain reaction (RT-PCR) coupled with direct cycling sequencing was developed and applied to seven patients.13 Subsequently, PCR-based restriction enzyme assays were developed to analyze nine single nucleotide polymorphisms (SNPs) in the IDUA gene. In the present study, we summarized the mutations found from 22 unrelated MPS I patients and calculated the frequencies of nine SNP alleles among normal American Caucasian and African-American populations.
PATIENTS AND METHODS
Patients and normal controls
Twenty-two patients from different regions of the United States were referred for mutation detection. All patients were enzymatically diagnosed by the demonstration of deficient IDUA activity in leukocyte, fibroblasts, or cultured trophoblasts.14 Whenever possible, clinical information and family data were obtained to evaluate the phenotypes and counsel for carrier detection. Peripheral blood samples or cultured fibroblasts or cultured trophoblasts from the affected individuals and peripheral blood samples from participating relatives were obtained. Peripheral blood samples from 56 North American Caucasians (33 males and 23 females) and 55 African Americans (24 males and 31 females) were collected as normal controls for SNP analysis. This study was approved by the institutional review board.
Detection of IDUA gene mutations in MPS I patients
From the collected peripheral blood cells, cultured fibroblasts, or cultured trophoblasts, total cellular RNA was extracted using the Trizol protocol (Fisher Scientific, Suwanee, GA) and genomic DNA was extracted using the Puregene DNA isolation kit (Gentra System, Minneapolis, MN). RT-PCR sequencing of cDNA fragments and PCR sequencing of genomic fragments were performed as previously described.13 Mutations detected from sequencing were further confirmed by a repeated PCR sequencing or a restriction enzyme assay provided that the mutations altered a restriction enzyme recognition site. Identified novel missense mutations were subjected to further screening on 100 normal chromosomes to rule out a polymorphism. The designation of detected mutations followed the suggested nomenclature previously reported.15
Polymorphic analysis of IDUA SNP alleles in control populations
The IDUA genomic sequence has designated a 1.8 kb segment A of exons 1,2 and intron 1 at its 5′ end, and a 4.5 kb segment B containing exons 3 to 14 and introns 3 to 13 at its 3′ end. The two segments are separated by a large intron 2 of approximately 15 kb in length.4 Polymorphism analyses were performed on nine IDUA intragenic SNPs. Three polymorphic alleles, A8(112C/A), A20(148G/A), and Q33H(187G/T), were located in segment A, and the other six, L118(440T/C), N181(631T/C), A314(1030G/C), A361T(1169G/A), T388 (1252G/C), and T410(1318C/G), were located in segment B. Genomic DNA samples from normal controls were subjected to PCR amplification using synthesized oligonucleotide primers flanking each SNP, then the amplicons were digested by selected restriction enzymes (Table 1). After completion of digestion, the reaction mixtures were loaded onto 2% to 3% agarose gel and fractionated by electrophoresis to detect the normal and polymorphic alleles. Allelic frequency for each SNP allele was determined by direct counting of the allele in the tested populations. The data were presented as percentages. For each SNP allele, the observed distribution of homozygous and heterozygous allelotypes was compared with the expectations from Hardy-Weinberg equilibrium by χ2 test. The constructs and frequencies of SNP haplotypes were estimated by a Bayesian haplotype inference algorithm with the use of a Haplotyper program.16
RESULTS
Mutational heterogeneity in MPS I patients
Of the 22 MPS I patients, 17 were fully genotyped and 5 were identified with only one mutation. Eleven different mutations including four nonsense mutations (Q70X, W402X, R621X, and R628X), five missense mutations (R162I, G208D, D315Y, P533R, and S633L), and two small deletions (1352delG, 1952del25bp) were identified. Table 2 summarizes the defined genotypes and single mutant allele, detected SNP alleles, and recognized phenotypes among the patients. The most frequent disease-causing mutations in our patient samples were the two previously described common mutations in the Caucasian population, W402X and Q70X,6 which accounted for approximately 39% (17/44) and 30% (13/44) of the total mutant alleles, respectively. The homozygotes and compound heterozygotes involving one and two of the common mutations were detected in 82% (18/22) of our patients. Mutations D315Y, P533R, R621X, R628X, and S633L were recurrent mutations previously reported in literature.5,6,17,18 Two novel missense mutations, R162I and G208D, were detected. Compound heterozygote R162I/R621X was found in cultured trophoblasts of an affected fetus. The G208D mutant allele was found in combination with the Q70X allele in one patient. A family study revealed the transmission of a paternal haplotype Q33H-Q70X and a maternal haplotype N181-G208D-A361T-T388 to the affected proband (Fig. 1). The novel mutation G208D is situated at the same codon as the previously described G208 V, which has been shown to be responsible for Hurler phenotype by clinical observation and in vitro cDNA expression assay.18 Two small deletions were detected. The 1352delG could cause a frameshift starting from codon 422 and resulted in a premature termination in codon 439. The 1952del25bp could cause a deletion of codons 622–629, a frameshift in subsequent coding, and an extended termination downstream of the normal termination codon.

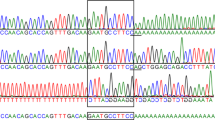
(A) Sequencing of PCR-amplified genomic fragments from a proband and relatives shows that the proband (II-2) is a compound heterozygote of mutations Q70X and G208D, the father (I-1) and sister (II-1) are carriers of mutation Q70X, and the mother (I-2) is a carrier of G208D. (B) Pedigree shows the transmission of paternal haplotype Q33H-Q70X and maternal haplotype N181-G208D-A361T-T388 to the proband.
For the 11 SNPs detected in the IDUA gene of the 22 patients studied, 7 (A8, A20, L118, N181, A314, T388, and T410) were silent mutations and four (Q33H, A361T, R105Q, and V454I) were missense mutations (Table 2). The Q33H-Q70X haplotype was observed in one case with homozygote Q70X and in six cases of compound heterozygotes involving the Q70X and other mutations. Another haplotype, L118-W402X, was observed in one case with homozygote W402X and in three cases with compound heterozygotes involving the W402X and other mutations. The L118 allele was previously reported as Kpn I site,9 and its association with a major MPS I mutation was suggested by linkage disequilibrium.19 Our data suggest that Q33H-Q70X and L118-W402X might be two representative haplotypes in Caucasian MPS I patients.
SNP frequency and haplotype inference in the IDUA gene
PCR-based restriction enzyme assay was performed on nine IDUA intragenic SNPs. The alleles that differ from the published sequences were denoted as the polymorphic alleles. The allelic frequency of each SNP allele was calculated as the percentage of polymorphic allele in the given total number of polymorphic and normal alleles. The cDNA locations, detected allelotypes, and calculated allelic frequencies of the nine SNPs are summarized in Table 3. The observed distribution of homozygous and heterozygous allelotypes was compared with the expectations from the Hardy-Weinberg equilibrium by χ2 test. The results indicate that the observed allelic frequencies were in good fitness with Hardy-Weinberg equilibrium (P values ranged from 0.2–0.96 for all alleles in Caucasians and 0.44–0.99 for alleles in African Americans). One exception was that of Q33H in African American controls (P value of 0.02). Differences in allelic frequencies for the nine SNPs between American Caucasians and African Americans were observed. The allelic frequencies from our samples were also different from those previously reported from an Australian population.9 This might be explained by the inherent differences between the tested populations that were from different geographic regions. In the present study, we observed the simultaneous occurrence of a similar type of allelotype in the A8 and A20 of IDUA gene segment A and in the N181, A314, A361T, T388, and T410 of segment B. Three major allelotype patterns of A8-A20 pairwise at segment A, A1{C/C-G/G}, A2{C/A-G/A}, and A3{A/A-A/A}, were observed in 32%, 35%, and 19% of Caucasian controls, respectively, and 44%, 43%, and 9% of African-American controls, respectively. Allelotype associations in N181-A314-A361-T388-T410 at segment B showed two major patterns, B1 {T/T-G/G-G/G-G/G-C/C} and B2 {T/C-G/C-G/A-G/C-C/G}. The two patterns B1 and B2 accounted for 75% and 13% in Caucasians, respectively, and 69% and 24% in African Americans, respectively. The results suggest the existence of two common haplotype structures C-G and A-A at A8-A20 in segment A, and also two common haplotype structures T-G-G-G-C and C-C-A-C-G at N181-A314-A361-T388-T410 in segment B. Haplotypes inferred from the allelotype data were constructed by a Haplotyper program, which uses a Bayesian Monte Carlo method with the underlying statistical model similar to that of expectation-maximization algorithm.16 The results indicate the presence of 15 haplotypes in the tested African-American samples and 17 haplotypes in the Caucasian samples. The two common haplotypes at A8-A20 pair, C-G and A-A, had frequencies of 54% and 38% in Caucasian controls, respectively, and 67% and 31% in African-Americans, respectively. The frequencies of the two common haplotypes at N181-A314-A361-T388-T410, T-G-G-G-C, and C-C-A-C-G were 84% and 6% in Caucasian controls, respectively, and 84% and 12% in African-Americans, respectively. Figure 2 A shows the constructs and frequencies of 12 major haplotypes (H1 to H12). Haplotypes with frequency <1% in both tested populations were considered rare variations and not included in the figure.

Polymorphism alleles and disease-causing mutations in the IDUA gene. (A) The constructs and frequencies of major haplotypes inferred from the Bayesian Haplotyper program. The number of chromosomes analyzed was 112 for Caucasian controls and 110 for African Americans. Black box and gray box denote the two common haplotypes in segments A and B of the IDUA gene, respectively. (B) characterized homozygous mutations and associated clinical phenotype (Ψ denotes pseudo allele).
DISCUSSION
The IDUA gene is divided into 14 exons spanning approximately 19 kb on human chromosome 4p16.3.4 PCR-based DNA sequencing has been a widely used approach for mutation detection in patients with MPS I.5,6 Experience from our laboratory and from another laboratory has demonstrated some limitations of this approach.6,18 In five cases, we could not identify a second mutation. Several explanations for the detection failure have been suggested. The major concerns include the insufficient sensitivity of the detection methods, the nature of IDUA gene content, and the variations of mutations. The RT-PCR-based method could likely miss some large deletions, rearrangements, and mutations in intronic or regulatory sequences, or mutations causing unstable and reduced amount of IDUA mRNA. The high GC content of the IDUA gene makes it possible that a mutation could be missed because of GC compressions despite efforts to sequence both cDNA and genomic amplicons. The existence of multiple polymorphic alleles in the IDUA gene also introduces extra effort in distinguishing disease-causing mutations from neutral polymorphisms.6,9,19,20 The following is a compiled list of intragenic SNP loci: A8(C/A), A20(G/A), Q33H(G/T), R105Q(G/A), G116R(G/A), L118(T/C), N181(T/C), A314(G/C), A361T(G/A), T388(G/C), T410(C/G), V454I(G/A), and R489(C/T). All of the SNPs, except Q33H, L118, and R489, occur in CpG dinucleotides and, therefore, are suggested to be hotspots for in situ methylation-deamination process. Nevertheless, our experience from mutation analysis of the 22 MPS I patients indicates that approximately 90% of mutant alleles will be detected by the PCR-based approach. Further analysis of the intronic and regulatory sequences of the IDUA gene will be required to increase the detection rate.
Several studies have described mutations in the IDUA gene. To date, a compilation of 39 nucleotide substitutions (missense/nonsense), 4 splicing mutations, 7 small deletions, 6 small insertions, and 1 small insertion/deletion in the IDUA gene are listed in the Human Gene Mutation Database.21 The present report will add four novel mutations to the mutation spectrum of the IDUA gene. The diversity of disease-causing mutations and the distribution of multiple polymorphic alleles are two characteristics of the human IDUA gene. In our patient samples, the two common mutations W402X and Q70X accounted for about 39% and 30% of the total mutant alleles and involved about 80% of patient genotypes. This result is consistent with the previous reports that the two are the most common mutations in Caucasian populations.6 Therefore, a rapid method of detecting an IDUA mutation profile consisting of the two common mutations and other frequent recurrent mutations could be developed for routine screening among Caucasian patients.
One important feature of the IDUA gene mutations is the variety of common mutations and the variations of relative frequency of these mutations in different ethnic populations at different geographic regions. IDUA gene mutations identified from Japanese,7 Arab,22 and Chinese patients20,23 show a different mutational spectrum from those detected in Caucasians.6,24 The relative frequency of common mutations and frequent recurrent mutations show obvious geographic heterogeneity.6–8,25–28 Table 4 lists the relative frequencies and clinical impacts of major IDUA mutations in different ethnic groups and geographic regions. The considerable mutational and geographic heterogeneity indicates that mutations in the IDUA gene might result from an independent mutagenesis process occurring in different populations at different times. Another important feature for the IDUA gene is the presence of multiple SNPs and possible major haplotype structures. Our observation on the possible linkage between Q33H and Q70X as well as L118 and W402X suggests the separate origins for the two common mutations in the Caucasian population. This result is consistent with previous reports that the two common mutations are associated with different haplotypes.17,24 The inferred haplotypes of A8-A20 and N181-A314-A361T-T388-T410 in our data require further confirmation from study of a larger sample population and assay of familiar haplotype segregation. It seems that the presence of two major haplotype structures C-G and A-A in segment A for A8-A20, and T-G-G-G-C and C-C-A-C-G in segment B for N181-A314-A361T-T388-T410 may require a simultaneous mutagenesis process and an allelic maintenance mechanism. This raises interesting questions about the mutagenesis mechanism and functional implications of these SNP alleles.
The ultimate goal of mutation analysis is to establish a genotype-phenotype correlation to provide a more accurate diagnosis of clinical subtypes and allow for improved prediction of disease progression. The combination of heterogeneous mutations in compound heterozygotes and the presence of multiple IDUA polymorphic alleles have made it difficult to assess the mutational effects. However, genotype-phenotype correlation observed from patients with homozygous mutant genotypes substantiated by functional assays such as enzyme kinetics or transfectional cDNA expression would be more reliable. Figure 2 B shows the recognized genotype-phenotype correlation for cases with homozygous mutations and functional assays by review of literature.12,18,20,24,25,29,30 A pseudodeficient allele identified in a compound heterozygote with W402X was also included.31 A general conclusion is that the IDUA gene has four sets of sequence variations: (1) null mutations and mutations causing coding disruption in a homozygous setting are most likely related to severe phenotype, (2) missense mutations result in a wide spectrum of clinical phenotypes ranging from severe to mild and require extensive study to characterize the functional impact, (3) pseudodeficient allele is rare and little is known about its effect on enzyme activity, and (4) polymorphic alleles are present as genetic background but may have minimal or no impact on phenotypes. Different combinations of the first three sets of mutations in compound heterozygotes contribute to the spectrum of clinical heterogeneity in the MPS I patients.
References
OMIM (Online Mendelian inheritance in man) Baltimore: Johns Hopkins University, Center for Medical Genetics, 1996. http://www.ncbi.nlm.nih.gov/omim.
Neufeld EF, Muenzer J . The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 1995: 2465–2494.
Scott HS, Anson DA, Orsborn AM, Nelson PV, Clements PR, Morris CP, Hopwood JJ . Human α-l-iduronidase: cDNA isolation and expression. Proc Natl Acad Sci U S A 1991; 88: 695–9699.
Scott HS, Guo XH, Hopwood JJ, Morris CP . Structure and sequence of the human α-l-iduronidase gene. Genomics 1992; 13: 1311–1313.
Scott HS, Bunge S, Gal A, Clarke LA, Morris CP, Hopwood JJ . Molecular genetics of mucopolysaccharidosis type I: diagnostic, clinical, and biological implications. Hum Mutat 1995; 6: 288–302.
Bunge S, Kleijer WJ, Steglich C, Beck M, Zuther C, Morris CP, Schwinger E, Hopwood JJ, Scott HS, Gal A . Mucopolysaccharidosis type I: identification of 8 novel mutations and determination of the frequency of the two common α-l-iduronidase mutations (W402X and Q70X) among European patients. Hum Mol Genet 1994; 3: 861–866.
Yamagishi A, Tomatsu S, Fukuda S, Uchiyama A, Shimozawa N, Suzuki Y, Kondo N, Sukegawa K, Orii T . Mucopolysaccharidosis type I: identification of common mutations that cause Hurler and Scheie syndrome in Japanese populations. Hum Mutat 1996; 7: 23–29.
Alif N, Hess K, Straczek J, Sebbar S, N'Bou A, Nabet P, Dousset B . Mucopolysaccharidosis type I: characterization of a common mutation that causes Hurler syndrome in Moroccan subjects. Ann Hum Genet 1999; 63: 9–16.
Scott HS, Nelson PV, Litjens T, Hopwood JJ, Morris CP . Multiple polymorphisms within the α-l-iduronidase gene (IDUA): implication for a role in modification of MPS-I disease phenotype. Hum Mol Genet 1993; 2: 1471–1473.
Lee-Chen GJ, Wang CK, Huang SF, Day KR . Human α-l-iduronidase (IDUA) gene: correlation of polymorphic DNA haplotypes and IDUA activity in Chinese population. Proc Natl Sci Counc Repub China B 1998; 22: 31–38.
Ashton LJ, Brooks DA, McCourt PAG, Muller VJ, Clements PR, Hopwood JJ . Immunoquantification and enzyme kinetics of α-l-iduronidase in cultured fibroblasts from normal controls and mucopolysaccharidosis type I patients. Am J Hum Genet 1992; 50: 787–794.
Bunge S, Clements PR, Byers S, Kleijer WJ, Brooks DA, Hopwood JJ . Genotype-phenotype correlations in mucopolysaccharidosis type I using enzyme kinetics, immunoquantification and in vitro turnover studies. Biochim Biophys Acta 1998; 1407: 249–256.
Li P, Thompson JN . Mutational analysis in seven patients with mucopolysaccharidosis type I [abstract]. Am J Hum Genet 1997; 61: A1490.
Thompson JN, Nowakowski RW . Enzymatic diagnosis of selected mucopolysaccharidoses: Hunter, Morquio type A, and Sanfillip types A, B, C, and D, and procedures for measurement of 35SO4-glycosaminoglycans. In: Hommes FA, editor. Techniques in diagnostic human biochemical genetics: a laboratory manual. New York: Wiley-Liss, 1991: 567–586.
Beaudet AL, Tsui LC . A suggested nomenclature for designating mutations. Hum Mutat 1993; 2: 245–248.
Niu T, Qin ZS, Xu X, Liu JS . Bayesian haplotype inference for multiple linked single-nucleotide polymorphisms. Am J Hum Genet 2002; 70: 157–169.
Scott HS, Litjens T, Nelson PV, Brooks DA, Hopwood JJ, Morris CP . α-l-Iduronidase mutations (Q70X, and P533R) associate with a severe Hurler phenotype. Hum Mutat 1992; 1: 333–339.
Beesley CE, Meaney CA, Greenland G, Adams V, Vellodi A, Young EP, Winchester BG . Mutational analysis of 85 mucopolysaccharidosis type I families: frequency of known mutations, identification of 17 novel mutations and in vitro expression of missense mutations. Hum Genet 2001; 109: 503–511.
Scott HS, Nelson PV, Cooper A, Wraith JE, Hopwood JJ, Morris CP . Mucopolysaccharidosis type I (Hurler syndrome): linkage disequilibrium indicates the presence of a major allele. Hum Genet 1992; 88: 701–702.
Lee-Chen GJ, Wang TR . Mucopolysaccharidosis type I: identification of novel mutations that cause Hurler/Scheie syndrome in Chinese families. J Med Genet 1997; 34: 939–941.
Krawczak M, Cooper DN . The Human Gene Mutation Database. Trends Genet 1997; 12: 121–122. http://www.uwcm.ac.uk/uwcm/mg/ns/.
Bach G, Moskowita SM, Tieu PT, Matynia A, Neufeld EF . Molecular analysis of Hurler syndrome in Druze and Muslim Arab patients in Israel: multiple allelic mutations of the IDUA gene in a small geographic area. Am J Hum Genet 1993; 53: 330–338.
Lee-Chen GJ, Lin SP, Tang YF, Chin YW . Mucopolysaccharidosis type I: characterization of novel mutations affecting alpha-l-iduronidase activity. Clin Genet 1990; 56: 66–70.
Clarke LA, Nelson PV, Warrington CL, Morris CP, Hopwood JJ, Scott HS . Mutation analysis of 19 North American mucopolysaccharidosis type I patients: identification of two additional frequent mutations. Hum Mutat 1994; 3: 275–282.
Gatti R, DiNatale P, Villani GR, Filocamo M, Muller V, Guo XH, Nelson PV, Scott HS, Hopwood JJ . Mutations among Italian mucopolysaccharidosis type I patients. J Inherit Metab Dis 1997; 20: 803–806.
Gort L, Chabas A, Coll MJ . Analysis of five mutations in 20 mucopolysaccharidosis type I patients: high prevalence of the W402X mutation. Hum Mutat 1998; 11: 322–323.
Voskoboeva EY, Krasnopolskaya XD, Mirenburg TV, Weber B, Hopwood JJ . Molecular genetics of mucopolysaccharidosis type I: mutation analysis among the patients of the former Soviet Union. Mol Genet Metab 1998; 65: 174–180.
Matte U, Leistner S, Lima L, Schwartz I, Giugliani R . Unique frequency of known mutations in Brazilian MPS I patients. Am J Med Genet 2000; 90: 108–109.
Tieu PT, Bach G, Matynia A, Hwang M, Neufeld EF . Four novel mutations underlying mild or intermediate forms of alpha-l-iduronidase deficiency (MPS IS and MPS IH/S). Hum Mutat 1995; 6: 55–59.
Menon KP, Neufeld EF . Evidence for degradation of mRNA encoding alpha-l-iduronidase in Hurler fibroblasts with premature termination alleles. Cell Mol Biol 1994; 40: 999–1005.
Aronovich EL, Pan D, Whitley CB . Molecular genetic defect underlying alpha-l-iduronidase pseudodeficiency. Am J Hum Genet 1996; 58: 75–85.
Acknowledgements
The authors thank the physicians, patients, and family members who participated in this study; Paula Huffman for her expertise in performing IDUA enzyme assays; and Jun Liu and Zhaohui Qin, Harvard School of Public Health, for help with the haplotyping analysis. This work was supported in part by a National Institutes of Health training grant (T32-CA09159-25) to P.L.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Li, P., Wood, T. & Thompson, J. Diversity of mutations and distribution of single nucleotide polymorphic alleles in the human α-l-iduronidase (IDUA) gene. Genet Med 4, 420–426 (2002). https://doi.org/10.1097/00125817-200211000-00004
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/00125817-200211000-00004
Keywords
This article is cited by
-
Genetic testing of Mucopolysaccharidoses disease using multiplex PCR- based panels of STR markers: in silico analysis of novel mutations
Metabolic Brain Disease (2019)
-
Cas9-mediated allelic exchange repairs compound heterozygous recessive mutations in mice
Nature Biotechnology (2018)
-
Phenotype prediction for mucopolysaccharidosis type I by in silico analysis
Orphanet Journal of Rare Diseases (2017)
-
Report of 5 novel mutations of the α-L-iduronidase gene and comparison of Korean mutations in relation with those of Japan or China in patients with mucopolysaccharidosis I
BMC Medical Genetics (2016)
-
Maternal mosaicism for IDUA deletion clarifies recurrence risk in MPS I
Human Genome Variation (2016)
