
Abstract
Mucopolysaccharidosis, type II (MPS II, MIM 309900) is a severe lysosomal storage disease with multisystem involvement. There is one product approved by the FDA, an enzyme replacement therapy, based on a phase III trial in older, attenuated MPS II individuals. Guidance on treatment of MPS II is lacking, not only in general, but for specific clinical situations. A previous systematic evidence-based review of treatment for MPS II demonstrated insufficient strength in all data analyzed to create a definitive practice guideline based solely on published evidence. The American College of Medical Genetics and Genomics (ACMG) Therapeutics Committee conducted a Delphi study to generate an MPS II clinical practice resource of the treatment for these individuals for the genetics community, based on the evidence-based review and subsequent literature. This report describes the process, including consensus development and areas where consensus could not be obtained due to lack of quality evidence. Recommendations from the Delphi process were generated, and areas were highlighted that need further study to help guide clinical care of these individuals.
Similar content being viewed by others

INTRODUCTION
The typical, severe form of mucopolysaccharidosis, type II (MPS II, MIM 309900) was first described in two brothers by the Canadian physician Charles Hunter in 1917.1 The estimated incidence varies from 1/60,000 to 1/150,000, with reports of higher rates among Ashkenazi Jews.2 It is X-linked and predominately a disease of males, although in rare cases affected females occur through skewing of X-chromosome inactivation.3 Iduronate-2-sulfatase (IDS), the enzyme deficient in MPS II, catalyzes the removal of sulfate groups from glycosaminoglycans (GAGs). It is targeted to the lysosome by the mannose-6-phosphate system. Loss of enzyme activity causes accumulation of GAGs in tissues and increased excretion of their breakdown products dermatan and heparan sulfate in urine. Enzyme deficiency may be due to total lack of enzyme, but is more often from decreased production, decreased catalytic activity, or protein misfolding.4
The signs of MPS II become apparent between the ages of 2 and 4 years with coarsening facial features, short stature, skeletal deformities (dysostosis multiplex), joint stiffness, hepatosplenomegaly, and progressive cognitive deterioration. Multiple organs may be affected as GAGs accumulate over time. Delays in developmental skills are typically evident by age 2 years, with plateauing and decline by age 4 to 6 years. Chronic otitis media and conductive hearing loss is present in most and many patients require hearing aids. Umbilical and inguinal hernias are also common, often at initial presentation. Upper airway obstruction manifests with snoring and sleep apnea, and wheezing is noted due to obstructive pulmonary disease. Cardiac disease leading to congestive heart failure occurs from both valve thickening leading to regurgitation and stenosis, and myocardial dysfunction secondary to infiltration with GAGs. Death occurs from the cardiac or pulmonary disease in most by 10 to 15 years of age.5 There is a broad spectrum of MPS II from the typical severe form to an attenuated or very mild form, with significant heterogeneity between the extremes. Individuals with attenuated disease have minimal if any neurological deficit and live into adulthood but still exhibit skeletal, joint, airway, and cardiac disease. Roughly two thirds of individuals with MPS II have the severe form of the condition, with some genotype–phenotype correlation6,7,8 and general consistency of phenotype between siblings in families.9
Treatment for MPS II has generally been directed at treating symptoms. Only one product specifically for MPS II treatment has been approved by the US Food and Drug Administration (FDA). Idursulfase (Elaprase®) is an intravenous enzyme replacement therapy (ERT) for MPS II that has been licensed in the United States by the FDA since 2006. A phase II/III trial of this product in individuals with attenuated MPS II over the age of 8 who were cognitively intact demonstrated improvement in some somatic manifestations. Clinical trial endpoint improvements were noted on the six-minute walk test (6MWT) distance and forced vital capacity on pulmonary function tests (PFT).10,11 Early evidence from the Hunter Outcome Survey suggest life span may be increased by ERT.12
Controlled trials of idursulfase have not been conducted on individuals with severe MPS II. Despite the lack of known therapeutic efficacy, individuals with severe MPS II have been treated with ERT. Small case series reporting ERT for the severe form of the disease seem to confirm benefits for reduction in liver and spleen volumes, joint range of motion, and possibly improved growth velocities.13 It could be anticipated that some somatic manifestations will improve in this group but that cognitive manifestations would not be improved due to the inability of the enzyme product to cross the blood–brain barrier. A phase I/II trial on the use of intrathecal enzyme replacement in severe MPS to overcome this obstacle has recently been reported, demonstrating preliminary safety data, but unfortunately no conclusive improvement in cognition.14 Hematopoietic stem cell transplant (HSCT) has also been used in MPS II. Earlier studies did not show benefit and demonstrated poor safety, but later case series suggest it may be effective.15 However, no HSCT clinical trial has been performed for any form of the disorder. Few long-term studies have been published, either in attenuated or severe forms, for any intervention.
Guidance on how to treat MPS II is lacking, not only in general, but for specific clinical situations. Previous guidelines for management of MPS II have been based on informal expert opinion without systematic evidence-based review16,17,18 with one previous Delphi method review19 and one Cochrane review.20 A guideline on treatment of severe MPS II specifically has also been published, based on clinical experience alone.21 Recommendations have varied, with statements to “consider” ERT in all patients with MPS II, usually with several caveats.
ERT is expensive and its precise benefits are uncertain. Guidance on many aspects of therapeutic management are needed by the community, including when to initiate ERT, how early in life to start for maximal benefit, when home therapy should be initiated, when to stop therapy, what benefits are expected, how to assess if therapy is working, and what to do if it is not, among many other questions that remain unaddressed by extant studies or current guidelines. To address the lack of guidance, the American College of Medical Genetics and Genomics (ACMG) Therapeutics Committee attempted to examine these issues through a commissioned, independent, systematic evidence-based review of all available data on the treatment of MPS II. Importantly, this review demonstrated insufficient strength in all data analyzed to create a definitive practice guideline based solely on published evidence.22 The purpose of this project was to use the evidence-based review as a basis to undertake a Delphi process using experts from a variety of disciplines that care for patients with MPS II to develop a practice resource for the community, providing consensus-based recommendations where possible.
MATERIALS AND METHODS
We applied study design and methodologic criteria as specified previously for Delphi studies,23,24 including definition of an expert, panel size, number of rounds, and a priori definition of consensus. We chose to use number of rounds as our strategy rather than attempting to reach consensus on items given that the quality of the evidence available might not encourage consensus based specifically on the evidence available.
Creating the working group
The ACMG Therapeutics Committee engaged a subcommittee of three members. An initial set of clinical questions was created according to patient, interventions, comparator, and outcome (PICO) methodology, using the initial systematic evidence-based review as the source for statement derivation. This group solicited members of a workgroup that would form the Delphi members, a writing group, and two chairs, and included a Delphi content expert. Individuals who treat MPS II patients were approached to join the workgroup, selected to be a heterogeneous mix of specialties. Conflict of interests were assessed, and the proposal and members were approved by the ACMG Board of Directors.
Systematic evidence-based review methodology
We utilized the previous review,22 and further added literature published since up to the time of Delphi study initiation (December 2018) using the terms and headings from Bradley supplemental table S2, with dates added to include articles after their search date (ended 31 December 2015) to 1 December 2018:
(((((Mucopolysaccharidosis II[mh] OR Mucopolysaccharidos*[tw]) AND (enzyme replacement therap*[mh] OR ERT[tw] OR idursulfase[tw] OR Elaprase[tw] OR idursulfase beta[tw] OR Hunterase[tw] OR hematopoietic stem cell transplantation[mh] OR bone marrow transplantation[mh] OR cord blood stem cell transplantation[mh])) AND English[lang])) AND (“2016/01/01”[Date—Publication]: “2018/12/01”[Date—Publication]))
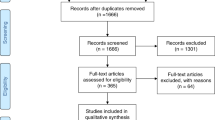
A total of 183 additional articles beyond those reviewed by Bradley et al. were retrieved in PubMed. Articles excluded from further review were (1) not MPS II patients, (2) case studies of 1–3 patients, (3) animal studies, or (4) review articles. The remaining articles (n = 29) were extracted to a collection and made available to the Delphi members through a shared online drive (Supplementary Material).
Delphi process
The Delphi group consisted of ten members. Two rounds were planned, with an optional third round if it was thought an additional round would generate additional consensus of statements. The Delphi members were provided access to the Bradley review and the subsequent articles that met their original inclusion criteria via an online shared drive, and members were specifically asked to use this information when rating the statements. Statements for the consensus process were created by the writing group from the initial evidence-based review. Surveys with these statements were created in REDCap and invitations sent to the Delphi group. Four weeks were allowed for completion of the survey, with two reminder emails sent prior to closing. A nine-point Likert scale was used to rate agreement or disagreement with the statement. A rating of 7 or more was defined as definite agreement, and a score of 3 or less as definite disagreement with the statement. A predefined threshold of 75% agreement or disagreement to a statement was used to indicate consensus. Participants were required to provide a comment if they did not agree with the statement, or if they wanted to provide feedback. Statements that did not reach consensus in round 1 were reviewed by the chairs. Based on respondent critiques and feedback, those statements needing clarification or division into separate statements were amended and sent out for round 2. Those that would not reach consensus due to lack of guiding data were not carried forward. Responses to round 1 were anonymized and shared with the Delphi members when the round 2 survey invitation was sent out.
Following data analysis and manuscript preparation, this document was reviewed and approved by the ACMG Board of Directors.
RESULTS
A total of eight Delphi members responded in round 1 and nine in round 2, with one person not completing either round. Statements and responses have been grouped together by theme below. A summary of statement consensus results is in Table 1. The full set of 37 statements in round 1 and 24 statements in round 2, with detailed results and Delphi member comments to the statements are included in the Supplementary Material.
Considerations regarding treatment initiation
We first attempted to define in which circumstances therapy should be initiated. There was broad consensus for initiating ERT across a range of considerations. Consensus was achieved for starting ERT for any age with signs or symptoms of severe phenotype or predicted to have severe disease by genotype of any age. Those individuals with a genotype predicted to be attenuated or with a genotype that could not predict phenotype had consensus for starting ERT if they were symptomatic or had signs of disease, but not if they did not have signs or symptoms of disease. Comments from the Delphi members debated what would constitute signs and symptoms, but broad consensus for initiating ERT was achieved using the unqualified statement. Consensus was also reached for the use of pressure equalizing (PE) tubes and hearing aids. No consensus for use of HSCT or intrathecal ERT (IT-ERT) could be reached, regardless of clinical circumstances considered.
A focused consensus emerged regarding use of ERT home therapy. Although the Delphi members provided comments on the importance of moving ERT infusions to the home, the comments were tempered with caution. This guided us to the creation of the statement “I would transition individuals to home therapy with early disease, minimal or easily controlled infusion reactions, and stable home” for which consensus could be reached.
Considerations regarding discontinuing therapy
We next attempted to define stopping points for ERT. We explored if no response to therapy should lead to discontinuation or if adverse reactions to therapy should require stopping. Various factors were explored, including length of time before deciding nonresponse (out to 18 months), presence of antibodies to idursulfase, or allergic reactions (including ability to ameliorate the reactions). No consensus could be obtained for any stopping rule explored, with the exception regarding ERT for an individual with severe MPS II and allergic reaction to ERT that could not be controlled by treatment. Comments from the Delphi members expressed reluctance to stop, even if there was no response. Many also felt that any reaction could be managed by simple measures (antihistamines, steroids, antipyretics) or even use of immunomodulation, thus stopping for adverse reactions was considered to be a rare circumstance.
Considerations regarding follow up and monitoring of therapy
Finally, we explored what types of follow up are useful for those on therapy. Consensus was reached for use of clinical exam for liver size, PFTs if they could be performed reliably, antibody testing for evaluating allergic reactions to ERT, urine GAGs, and neuropsychology testing. Consensus was not reached for use of the 6MWT (other than to exclude use if reliability was a concern), diagnostic imaging of liver or spleen size, and routine annual magnetic resonance image (MRI) of the neck.
DISCUSSION
Most rare diseases, even those with approved therapies, lack enough high-quality data to be able to create an evidence-based guideline to assist clinicians in the care of these individuals. MPS II is no exception. Our attempts to gather all available data from multiple sources, including gray sources (material produced by organizations or government outside of academic publishing), via a systematic evidence-based review failed to yield enough information to create a formal clinical guideline based on evidence alone. We attempted to provide an alternative means for guidance through an unbiased expert consensus statement using a Delphi approach.
The process demonstrated the difficulty of establishing guidance when few data are available. The Delphi members were able to reach consensus on statements for which there were good clinical data; however, for clinical questions that had minimal or conflicting published information, expert opinions reflected the uncertainty. One example is the key clinical question of not only when and on whom to initiate ERT, but also when it should be discontinued. Consensus was reached regarding initiation for those with the severe form of MPS II and those with attenuated MPS II who showed signs or symptoms of disease. Our expert panel was unable to establish a consensus on the critical clinical decision to stop therapy outside of allergic reaction that could not be controlled, as no information exists on discontinuation in the literature. Either evidence-based or consensus-based decisions about termination of therapy would depend on a better definition of treatment utility. Some therapies were also difficult to evaluate with no consensus developed for use of IT-ERT and HSCT. Although consensus could not be reached, these additional therapies could still be appropriate interventions if more information accumulates, particularly on HSCT and IT-ERT. Of note, although PFTs and 6MWT were used as defining endpoints in the only phase III trial for ERT, most of our Delphi members did not feel these were very useful measures for following patients, with comments in particular about futility in using them for severe MPS II. It is thus important that better measures be established for defining success in outcomes, particularly as further analysis and additional clinical trials are undertaken.
Our Delphi study yields the following recommendations:
1. All individuals with severe MPS II or predicted to have severe MPS II based on genotype warrant starting ERT, prior to showing signs or symptoms.
2. Individuals with signs or symptoms with either attenuated or severe MPS II warrant ERT.
3. Individuals with attenuated MPS II who are not showing signs or symptoms of disease do not warrant ERT.
4. Home infusions may be considered for those with early disease, easily managed ERT infusion reactions, and a stable home environment.
5. Individuals receiving ERT who have developed allergic reactions that cannot be controlled by standard therapies or immunomodulation should have ERT discontinued.
6. PE tubes and hearing aids are useful therapies.
7. Clinical evaluation of liver and spleen size are recommended for judging clinical effectiveness of treatment, with optional use of imaging modalities (ultrasound or MRI of the abdomen) to follow organ size. PFTs are recommended if the individual can reliably perform them, but there are concerns on the utility of the 6MWT. Lab studies of GAGs are recommended, as well as antibodies to ERT to assess infusion reactions. Finally, neuropsychology testing is recommended for following disease progress.
The statements reaching and not reaching consensus differ from previous expert opinion guidelines.16,17,19,21 There is broad agreement across studies with ours on considering treatment for symptomatic individuals with ERT. We differ for those predicted to be severe prior to onset of signs and symptoms, recommending ERT. Compared with Latin America guidelines16 that do not suggest use under age 6, we consider all ages eligible. Guidelines differ regarding discontinuation of therapy. Our study was not able to define a set of stopping rules, whereas previous guidelines have recommended stopping if no effect is noted after 6–12 months of ERT and stopping ERT near the end of life.21 Our study and previous expert opinions are similar regarding evaluation and follow-up recommendations, reflecting pragmatism, as instruments used in the phase III trial are not easily transferred to the clinical setting for use in younger and more severely affected MPS II individuals.
Our study highlights the difficulties in the field of rare disease therapeutics to assemble evidence and create guidance documents for clinicians. Numerous rare diseases have not had any phase III trials to evaluate therapies, nor may they be possible in many circumstances. Narrow scopes of phase III clinical trials, while establishing short term efficacy and safety in small select populations, will not provide sufficient information to guide all aspects of clinical care. Postmarketing follow-up studies (phase IV patient registries such as the Hunter Outcome Survey25) may fill in some gaps, but still leave many questions. The balance between bringing needed therapies to market for individuals and families with these severe and lethal conditions and generating enough evidence in a limited number of affected individuals to fully inform clinicians will continue to pose problems. In addition, evidence-based reviews are unlikely to find enough data for clinical practice guidelines that meet minimal criteria laid out by the Institute of Medicine,26 leaving imperfect expert consensus methods as the best approach to create guidance for clinicians.
We attempted to make our process as transparent as possible and to use an expert group as heterogeneous as ACMG policies would allow. This did create limitations to our process, with a smaller Delphi group size that is limited in the number of non-ACMG members permitted and did not allow us to include patient advocates.
Future research should address the major deficiencies identified here: When is initiation of ERT not warranted? What should guide the clinician to stop ERT? Given recent literature on the use of immune modulating therapy in Pompe syndrome,27,28 should immune tolerance induction also be considered for ERT in Hunter syndrome? What regimen should be used, and in which setting—prophylactic, after development of neutralizing antibodies, only for certain genetic variants? How does HSCT compare with ERT in a clinical trial? In addition, what do new therapies under study (IT-ERT, gene therapies) offer and when should they be considered? What is the best way to assess success of therapy—do we need new clinical evaluations, patient and family important endpoints, or better biomarkers? As more information becomes available, we hope to repeat our process to give better resources to the genetics community.
References
Hunter C. A rare disease in two brothers. Proc R Soc Med. 1917;10:104–116.
Nelson J, Crowhurst J, Carey B, Greed L. Incidence of the mucopolysaccharidoses in Western Australia. Am J Med Genet A. 2003;123A:310–313.
Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Valle D, Antonarakis S, Ballabio A, Beaudet A, Mitchell GA, editors. The online metabolic and molecular bases of inherited disease. New York: McGraw-Hill Education; 2019.
Froissart R, Da Silva IM, Maire I. Mucopolysaccharidosis type II: an update on mutation spectrum. Acta Paediatr. 2007;96:71–77.
Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J. Initial report from the Hunter Outcome Survey. Genet Med. 2008;10:508–516.
Dvorakova L, Vlaskova H, Sarajlija A, et al. Genotype-phenotype correlation in 44 Czech, Slovak, Croatian and Serbian patients with mucopolysaccharidosis type II. Clin Genet. 2017;91:787–796.
Saito S, Ohno K, Okuyama T, Sakuraba H. Structural basis of mucopolysaccharidosis type II and construction of a database of mutant iduronate 2-sulfatases. PLoS One. 2016;11:e0163964.
Vollebregt AAM, Hoogeveen-Westerveld M, Kroos MA, et al. Genotype-phenotype relationship in mucopolysaccharidosis II: predictive power of IDS variants for the neuronopathic phenotype. Dev Med Child Neurol. 2017;59:1063–1070.
Ficicioglu C, Giugliani R, Harmatz P, Mendelsohn NJ, Jego V, Parini R. Intrafamilial variability in the clinical manifestations of mucopolysaccharidosis type II: data from the Hunter Outcome Survey (HOS). Am J Med Genet A. 2018;176:301–310.
Muenzer J, Gucsavas-Calikoglu M, McCandless SE, Schuetz TJ, Kimura A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol Genet Metab. 2007;90:329–337.
Muenzer J, Wraith JE, Beck M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med. 2006;8:465–473.
Burton BK, Jego V, Mikl J, Jones SA. Survival in idursulfase-treated and untreated patients with mucopolysaccharidosis type II: data from the Hunter Outcome Survey (HOS). J Inherit Metab Dis. 2017;40:867–874.
Lampe C, Bosserhoff AK, Burton BK, et al. Long-term experience with enzyme replacement therapy (ERT) in MPS II patients with a severe phenotype: an international case series. J Inherit Metab Dis. 2014;37:823–829.
Muenzer J, Hendriksz CJ, Fan Z, et al. A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysaccharidosis II. Genet Med. 2016;18:73–81.
Kubaski F, Yabe H, Suzuki Y, et al. Hematopoietic stem cell transplantation for patients with mucopolysaccharidosis II. Biol Blood Marrow Transplant. 2017;23:1795–1803.
Giugliani R, Villarreal ML, Valdez CA, et al. Guidelines for diagnosis and treatment of Hunter syndrome for clinicians in Latin America. Genet Mol Biol. 2014;37:315–329.
Muenzer J, Beck M, Eng CM, et al. Multidisciplinary management of Hunter syndrome. Pediatrics. 2009;124:e1228–e1239.
Scarpa M, Almassy Z, Beck M, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72.
Gonzalez-Gutierrez-Solana L, Guillen-Navarro E, Del Toro M, Dalmau J, Gonzalez-Meneses A, Couce ML. Diagnosis and follow-up of patients with Hunter syndrome in Spain: a Delphi consensus. Medicine (Baltimore). 2018;97:e11246.
da Silva EM, Strufaldi MW, Andriolo RB, Silva LA. Enzyme replacement therapy with idursulfase for mucopolysaccharidosis type II (Hunter syndrome). Cochrane Database Syst Rev. 2016;2:CD008185.
Muenzer J, Bodamer O, Burton B, et al. The role of enzyme replacement therapy in severe Hunter syndrome-—an expert panel consensus. Eur J Pediatr. 2012;171:181–188.
Bradley LA, Haddow HRM, Palomaki GE. Treatment of mucopolysaccharidosis type II (Hunter syndrome): results from a systematic evidence review. Genet Med. 2017;19:1187–1201.
Diamond IR, Grant RC, Feldman BM, et al. Defining consensus: a systematic review recommends methodologic criteria for reporting of Delphi studies. J Clin Epidemiol. 2014;67:401–409.
Waggoner J, Carline JD, Durning SJ. Is there a consensus on consensus methodology? Descriptions and recommendations for future consensus research. Acad Med. 2016;91:663–668.
Muenzer J, Jones SA, Tylki-Szymanska A, et al. Ten years of the Hunter Outcome Survey (HOS): insights, achievements, and lessons learned from a global patient registry. Orphanet J Rare Dis. 2017;12:82.
Institute of Medicine. Clinical practice guidelines we can trust. Washington, DC: The National Academies Press; 2011.
Banugaria SG, Prater SN, Patel TT, et al. Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic material-negative classic infantile Pompe disease: a step towards improving the efficacy of ERT. PLoS ONE. 2013;8:e67052.
Doerfler PA, Nayak S, Corti M, Morel L, Herzog RW, Byrne BJ. Targeted approaches to induce immune tolerance for Pompe disease therapy. Mol Ther Methods Clin Dev. 2016;3:15053.
Acknowledgements
The MPS II Workgroup and ACMG Therapeutics Committee gratefully acknowledges the work of the Delphi members: Georgianne Arnold, Michael Bober, Barbara Burton, Maria Escolar, Nancy Leslie, Saadet Mercimek-Andrews, Lawrence Merritt, John Mitchell, Laurie Smith, and Reid Sutton.
Author information
Authors and Affiliations
Consortia
Ethics declarations
Disclosure
K.L.M. has received support for research from Abeona, Biomarin, Homology and Shire. Members of the Delphi group were reviewed by the Conflict of Interest Committee of the American College of Medical Genetics and Genomics. Conflicted members constituted less than half of the group, in accordance with existing bylaws, and no conflict precluded involvement in the workgroup. The other authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Disclaimer:
This practice resource is designed primarily as an educational resource for medical geneticists and other clinicians to help them provide quality medical services. Adherence to this practice resource is completely voluntary and does not necessarily assure a successful medical outcome. This practice resource should not be considered inclusive of all proper procedures and tests or exclusive of other procedures and tests that are reasonably directed to obtaining the same results. In determining the propriety of any specific procedure or test, the clinician should apply his or her own professional judgment to the specific clinical circumstances presented by the individual patient or specimen.
Clinicians are encouraged to document the reasons for the use of a particular procedure or test, whether or not it is in conformance with this practice resource. Clinicians also are advised to take notice of the date this practice resource was adopted, and to consider other medical and scientific information that becomes available after that date. It also would be prudent to consider whether intellectual property interests may restrict the performance of certain tests and other procedures.
The Board of Directors of the American College of Medical Genetics and Genomics approved this practice resource on 22 June 2020.
Supplementary information
Rights and permissions
About this article

Cite this article
McBride, K.L., Berry, S.A., Braverman, N. et al. Treatment of mucopolysaccharidosis type II (Hunter syndrome): a Delphi derived practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 22, 1735–1742 (2020). https://doi.org/10.1038/s41436-020-0909-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0909-z
Keywords
This article is cited by
-
Caregiver experiences and observations of intrathecal idursulfase-IT treatment in a phase 2/3 trial in pediatric patients with neuronopathic mucopolysaccharidosis II
Orphanet Journal of Rare Diseases (2024)
-
Clinical investigator perspectives on patient outcomes in children with neuronopathic mucopolysaccharidosis II during intrathecal idursulfase-IT treatment
Orphanet Journal of Rare Diseases (2024)
-
A post hoc analysis of Projected Retained Ability Scores (PRAS) for the longitudinal assessment of cognitive functioning in patients with neuronopathic mucopolysaccharidosis II receiving intrathecal idursulfase-IT
Orphanet Journal of Rare Diseases (2023)
