
Abstract
Objective
To retrospectively assess the clinicopathological characteristics of orbital granular cell tumours (GCTs).
Methods
A non-comparative review of the clinical characteristics, imaging, histopathological features, management, and prognosis of five cases of benign GCT and one case of malignant GCT (MGCT) was conducted, along with a review of the English language literature.
Results
Among the six cases, four tumours were adherent to the extraocular muscle (EOM), and three tumours to the optic nerve (ON). Morphologic examinations revealed polygonal cells containing periodic-acid-Schiff-positive eosinophilic granules. All tumours (100%) were positive for VIM and NSE, five (83.3%) tumours were positive for S-100, and three (50%) tumours were positive for CD68. The follow-up examination of the MGCT witnessed recurrence and brain metastasis despite several thorough resections, but the patient remained alive; the follow-up examination of the four benign GCTs that had received incomplete excision revealed recurrence in one patient and dramatic shrinkage of the residual tumour in another; there was no recurrence in the other two patients.
Conclusions
GCT should be considered in the differential diagnosis of orbital tumours, which may affect EOMs and ON. The natural course of GCT can include tumour progression, stability, or spontaneous regression. To avoid recurrence, complete resection is recommended for orbital GCT. To the best of our knowledge, primary orbital MGCT is reported for the first time.
Similar content being viewed by others

Introduction
Since they were first described by Abrikossoff in 1926, granular cell tumours (GCTs) have been reported in several regions of the body, mostly in the skin and subcutaneous tissue of the neck and head. GCT is a rare, usually benign, soft-tissue tumour that mainly affects adults during the fourth to sixth decades of life, with a female predilection. Malignant GCT (MGCT) is even rarer. The histogenesis of GCT, which rarely arises in the orbit, has been debated for decades. Although several cases of orbital GCTs have been reported in the literature,1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 to the best of our knowledge, no cases of primary orbital MGCT have yet been described.
The purpose of the present study was to analyse the clinicopathological features of orbital GCT. To achieve this goal, we examined six cases of orbital GCT that had been diagnosed and treated in the Eye and ENT Hospital of Fudan University, and also reviewed previous studies of orbital GCTs. We believe that this study constitutes the largest series of orbital GCTs ever reported. In addition, among the examined cases was one case of primary orbital MGCT and one case of non-neural orbital GCT, both of which (to our knowledge) are reported here for the first time.
Materials and methods
The study protocol was approved by the institutional review board of Eye and ENT Hospital of Fudan University, Shanghai, China. The medical records of patients who had been admitted to the Eye and ENT Hospital of Fudan University since 2001 were reviewed; six patients with orbital GCT were identified. We retrieved the patients’ demographic data, presentation, ocular examinations, orbital imagings, clinical course, and treatments from the records. Hematoxylin and eosin (HE) staining and immunohistochemistry for S-100, CD68, vimentin (VIM), neuron-specific enolase (NSE), periodic-acid-Schiff (PAS), creatine kinase (CK), and smooth muscle actin (SMA) were performed on tumour sections. Immunohistochemistry staining for HMB-45 and Melan-A was also conducted on MGCT tumour sections to distinguish them from malignant melanoma, which was also positive for S-100. Follow-ups were performed through clinic service or telephone interviews.
At the same time, a search of PubMed and Web of Science was conducted, using the following keywords: ‘Granular cell tumour’, ‘Granular cell myoblastoma’, and ‘Abrikossoff’s tumour’. Articles about GCT in the eye region and written in English were retrieved; the results returned articles with both the US spelling of ‘tumor’ and the British spelling of ‘tumour’. Intraocular tumours, and tumours found on the ocular surface or within the eyelid, were subsequently eliminated. The clinicopathological features and prognosis of the orbital GCTs were reviewed.
Results
Clinical characteristics
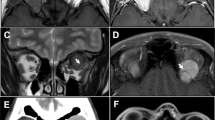
Our study included five cases of benign GCT and one case of MGCT. The clinical information is described in detail in Table 1. Computed tomography (CT) showed diffuse isodense masses, while magnetic resonance imaging (MRI) showed fusiform tumours, isointense to the extraocular muscles (EOMs) in T1- and T2-weighted imaging with contrast enhancement, as shown in Figures 1 and 2.

(a–c) Magnetic resonance imaging (MRI) and computed tomography (CT) scans from patient 3, obtained before surgery and at 17 months and 74 months after surgery, showing dramatic shrinkage of the orbital GCT. (d) The tumour cells were separated by fibrous tissue and arranged in a ribbon pattern (hematoxylin and eosin, × 50). (e) The cells had a characteristic granular eosinophilic cytoplasm with sporadic muscle fibres among them (arrowhead) (hematoxylin and eosin, × 400). (f) Strong and diffuse immunostaining for S-100 (× 50).

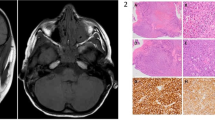
(a–c) CT scan of patient 4 obtained at 0 month, 29 months, and 51 months after diagnosis. (d) The tumour cell arrangement in MGCT was disordered (hematoxylin and eosin, × 50). (e) Relatively larger tumour cells with prominent cells, nuclear pleomorphism, and vesicular nuclei. Mitotic figures were present (arrowhead) (hematoxylin and eosin, × 400). (f) Strong and diffuse immunostaining for S-100 (× 50). (g) The immunonegativity for HMB-45 (× 200). (h) the immunonegativity for Melan-A (× 200).
Histopathology and immunohistochemistry
The histological sections stained with HE and S-100 are shown in Figures 1 (patient 3) and 2 (patient 4). In benign GCTs (patients 1–3, 5, and 6), the tumour cells were arranged in nests or ribbons, separated by fibrous tissue. There was sporadic infiltration of inflammatory cells in the peripheral regions, mostly composed of lymphocytes. The tumour cells were polygonal, with well-defined borders. Cytoplasm was abundant, with numerous typical eosinophilic granules. The nuclei were round with small nucleoli, although scattered cells with vesicular nuclei were also observed. No mitotic or necrotic areas could be found. The tumour cells presented an infiltrative growth pattern. Scattered striated muscle fibres were also present in patients 3 and 6, but were separate from the tumour cells. In the MGCT case (patient 4), the tumour cells presented with coarse granular eosinophilic cytoplasm resembling benign GCT, although the arrangement of the cells was disordered: the tumour cells were relatively larger, exhibiting prominent cell and nuclear pleomorphism, vesicular nuclei, and large nucleoli, and mitotic figures were present. All of these malignant changes were widespread in the whole tumour section, without any benign-appearing parts in the tumour. All of the specimens were positive for PAS, NSE, and VIM (Table 2); five (83%) were positive for S-100; and three (50%) were positive for CD68. The specimens were all negative for CK and SMA (data not shown); furthermore, the MGCT was negative for HMB-45 and Melan-A (Figure 2).
Review of orbital GCTs
We retrieved 29 articles describing 34 cases of orbital GCTs. The clinicopathological data are summarised in Tables 2 and 3; a total of 40 cases were analysed. Patients ranged from 4–75 years of age, with an average age of 44 years. Among the tumours, 38 were benign GCTs, 1 was a MGCT metastatic to the orbit, and 1 was primary MGCT (patient 4). It should be noted that GCTs were misdiagnosed as inflammatory conditions in the biopsy of patient 4 in our series, as well as in another two cases from the literature.11, 26 The most common presentations were diplopia (40%, 16/40) and proptosis (32.5%, 13/40). EOM involvement was described in 67.5% (27/40) of the tumours, of which the most affected EOM was the inferior rectus muscle (40.7%). Adhesion to the ON was reported in 32.5% (13/40) of cases, based on radiological and mid-surgical findings. In the 22 cases in which immunohistochemical studies were performed (Table 2), 100% (10/10) of cases available for VIM were positive, 90.9% (20/22) of cases were positive for S-100, 88.9% (8/9) of the cases available for NSE were positive, and 66.7% (6/9) of the cases available for CD68 were positive.
With the exception of one patient who died of cerebral dysregulation due to a GCT in the middle cranial fossa, and one patient for whom no treatment information was available, all of the patients were treated surgically. Follow-ups were conducted with 23 patients, with an average follow-up duration of 29 months (range: 1–96 months). The patients with MGCT presented unsatisfying prognosis, as the primary MGCT (patient 4) developed metastases, and the patient with metastatic orbital MGCT died of metastases.18 For the benign GCTs cases, 100% (13/13) of cases exhibited no recurrence when resected entirely; when they were resected subtotally, however, 37.5% (3/8) of the cases recurred, and 62.5% (5/8) of the patients displayed no recurrence, including one who showed regression of the residual tumour after incisional biopsy (patient 3).
Discussion
GCT was once thought to originate in the striated muscle because of its association with muscle tissue; this explains its previous name of ‘granular cell myoblastoma’. This theory has been challenged, however, by the presence of tumours in organs that lack striated muscle,30 the absence of glycogen in tumour cells, and the absence of any similarity between tumour cells and striated muscle under electron microscopy (EM).31 Researchers have subsequently proposed other cells of origin, including fibroblasts32 and primitive mesenchymal cells,10 as well as an origin resulting from metabolic derangement involving histiocytes.33 The idea of GCT derivation from Schwann cells has gained popularity recently, mainly on the basis of evidence that the cytoplasmic granules in Schwann cells are indistinguishable from those found in GCT cells.34
Clinical features
The most common complaints of orbital GCT patients are diplopia, proptosis, and mobility restriction. In CT scans, GCTs appear as diffuse or oval masses isodense relative to brain tissue. The tumours are isointense relative to EOMs in T1-weighted MRI and isointense or slightly hyperintense in T2-weighted MRI, which can be slightly enhanced with contrast administration. Surgical findings describe GCT as an oval mass that may infiltrate adjacent EOMs.
In the current study, EOM involvement was seen in 67.5% of cases. The inferior rectus muscle is most commonly affected (40.7%). The fact that neural-derived GCT is closely associated with EOMs may be due to the dense neural supply of EOMs.27
Vision decrease/loss due to tumour adhesion to the ON was noted in 32.5% of cases in our study. Strangely, the ON is thought to be an outgrowth of the central nervous system (CNS), which does not possess Schwann cells. Given this fact, it is fair to wonder why GCT so frequently appears to be associated with the ON. A few authors believe that the pathogenesis of GCT varies, based on the tumour’s location. Although a Schwann cell origin is favoured in most cases, an astrocytic origin has been proposed in the literature for GCTs in CNS locations; this suggestion is mainly based on the immunoreactivity of tumour cells to glial fibrillary acidic proteins.35 Despite the lack of Schwann cells in the ON, however, sympathetic nervous composition has been observed to be present in the meninges surrounding it. In our opinion, it is this sympathetic nervous composition—which contains Schwann cells—that may be responsible for the frequently observed association of GCT with the ON.
Pathologic and differential diagnosis
The majority of GCTs are benign. Histologically, GCT is composed of sheets or nests of round or polygonal cells with small and round nuclei;36 the most characteristic feature is abundant cytoplasm with eosinophilic granules, which are PAS positive. EM has revealed these granules to be secondary lysosomes.23 No mitotic figures or necrosis is present in these tumours; immunohistochemical assessment is needed for the differential diagnosis. The majority of orbital GCTs are positive for S-100, NSE, VIM, and CD68, but negative for epithelial, muscle, and melanin markers. It should be noted, however, that the presence of lymphocytes at the tumour periphery may lead to the misdiagnosis of tumours as an inflammatory condition, a phenomenon that was observed in three cases in our study. The implications of lymphocyte infiltration are not yet clear, but it may reflect an immunoresponse to the infiltrative pattern of the tumour. In our opinion, a larger tumour sample obtained by means of lateral orbitotomy is essential to allow correct diagnoses to be made when conducting biopsies.
Several diseases that resemble orbital GCT in pathology should be considered in the differential diagnosis: especially those that feature similar granular cytoplasm. Alveolar soft part sarcoma (ASPS) is characterised by distinctive organoid nests of polygonal tumour cells separated by thin-walled capillaries. The tumour cells display abundant PAS-positive intracytoplasmic crystals under light microscopy, and numerous mitochondria and an extensive Golgi complex under EM. Abundant PAS-positive granular cytoplasm can also be observed in embryonal rhabdomyosarcoma. This type of tumour can be differentiated by distinct cross-striations in the cytoplasm, however, by positivity for muscle stains such as desmin, MyoD1, and actin. Melanoma (another tumour of neural crest cell origin) may simulate GCT, but should be easily distinguishable from GCT with staining for HMB-45 and Melan-A.
Non-neural GCT
S-100 protein is an acidic protein in cells of neural crest origin, the expression of which is strongly supportive of a peripheral nerve sheath origin for GCTs. There are subsets of S-100-negative GCTs in skin,37, 38 which are termed ‘non-neural GCTs’. A few authors believe that these tumours are associated with an altered differentiation process in the MGCTs of Schwann cell origin,39 or with a primitive mesenchymal cell origin.40 The nuclear atypia and mitotic activity of non-neural GCTs are more obvious than those of conventional GCTs, which suggest malignant potential, but the clinical manifestations appear to be relatively benign.41 The current study includes two S-100-negative orbital GCTs that recurred following surgery, however, which is inconsistent with this idea. McNab et al14 reported a case of benign orbital GCT that was S-100 negative. In two years of follow-up after an incomplete excision, the tumour showed a small extension into the cavernous sinus through the superior orbital fissure. Patient 1 in our hospital also presented with an S-100 negative tumour, with no signs of malignancy in pathological studies. This patient experienced tumour recurrence, however, 4 months following a subtotal resection. Both of these cases may belong to the group of non-neural GCTs, thus contributing important information to the body of knowledge about this uncommon tumour type: specifically that orbital non-neural GCT can be consistent with conventional GCT in pathology, but the clinical course may be benign or could demonstrate malignant potential. Long-term follow-ups are necessary to obtain further information.
MGCTs
MGCTs are extremely rare, accounting for <2% of all GCTs. They can be categorised into clinically and pathologically malignant tumours: the former are histologically benign, but clinically malignant with metastasis, while the latter are malignant both histologically and clinically.42 The pathological differentiation of malignant from benign GCT has been a challenge for researchers. The most widely accepted diagnostic criteria comprise six histologic features: increased mitotic activity (>2 mitoses/10 high-power fields at × 200 magnification), necrosis, tumour cell spindling, vesicular nuclei with prominent nucleoli, increased nuclear/cytoplasmic ratio, and pleomorphism.43 The presence of three or more criteria is consistent with malignancy, while the presence of one to two defines an ‘atypical’ GCT. These features must also be present in the majority of the tumour, rather than focally.
Previously, only one ‘primary’ and one metastatic orbital MGCT had ever been reported in the literature. The ‘primary’ MGCT was described as a painless mass in the lower eyelid, with apparent generalised metastasis,1 although this tumour was later reclassified as an ASPS by Allaire et al.16 Callejo et al18 reported a case of a patient with orbital MGCT metastasising from the cervical region, who died (presumably of metastases) 4 years after the diagnosis of primary GCT. The aggressive clinical course and pathological findings of metastasis confirmed MGCT.
In this study, we have reported on a case of primary orbital MGCT that was both clinically and histologically malignant, exhibiting all six characteristics of malignancy in multiple slides. MGCTs, unlike benign tumours, demonstrate aggressive progression, with rapid growth and metastatic potential; 39% of patients were observed to die of disease within 3 years.43 Resection with adequate surgical margins is the preferred treatment, and neither chemotherapy nor radiotherapy has been proved useful for MGCTs. A dramatic response to pazopanib in a patient with MGCT metastatic to multiple regions was reported recently,44 which may provide a promising treatment.
Treatment and prognosis
Both complete removal and subtotal resection have been described for GCT in the published literature. Consistent with a previous report,27 we witnessed no recurrence following complete surgical removal. While McNab et al14 once reported that many tumours with incomplete excision also exhibited no recurrence, in our cases (and in our review of the literature) 37.5% of benign GCTs with subtotal resection demonstrated tumour recurrence. Because two of the three tumours were S-100 negative, we cannot determine whether the recurrence was completely due to subtotal resection or due to the tumours’ particular pathogenesis. All of the findings to date nonetheless continue to suggest to the ophthalmologist that complete resection should be considered as the treatment of choice for GCTs; given the frequent infiltration of these tumours to adjacent tissues, however, complete resection may be impossible. Under such circumstances, extended resection may be appropriate, especially in cases with destroyed function or potential progression. We were surprised to observe spontaneous regression in one case (patient 3) with subtotal resection, which, to our knowledge, is the first report in the literature of this happening.
The exact mechanisms that cause spontaneous regression remain unclear, and findings from tumour pathology show nothing special that could explain the phenomenon. It would be interesting to conduct further studies to determine whether tumour differentiation plays a role. This case also contributes to what is known about the natural course of GCTs, which might involve progression, stability, or spontaneous regression. It is a remarkable fact that, in cases in the literature review that involved EOMs, complete excision of the tumour did not provide optimal prognosis for motility in the majority (73.3%) of cases.27 Similarly, in cases involving the ON, surgery cannot improve the patient’s vision. The possibility of tumour infiltration into the ON should remind ophthalmologists to perform a comprehensive evaluation of the surgical results, and to inform patients properly.
In conclusion, orbital GCTs are usually benign; we have reported in this study the first case of a primary orbital MGCT with an unfavourable prognosis. GCT should be considered in the differential diagnosis of orbital tumours, especially when an EOM or the ON is affected. Complete resection is recommended to ensure that tumours will not recur.

References
Dunnington JH . Granular cell myoblastoma of the orbit. Arch Ophthal 1948; 40 (1): 14–22.
Morgan LR, Fryer MP . Granular cell myoblastoma of the eye. Case report. Plast Reconstr Surg 1969; 43 (3): 315–317.
Chaves E, Oliveira AM, Arnaud AC . Retrobulbar granular cell myoblastoma. Br J Ophthalmol 1972; 56 (11): 854–856.
Gonzalez-Almaraz G, de Buen S, Tsutsumi V . Granular cell tumor (myoblastoma) of the orbit. Am J Ophthalmol 1975; 79 (4): 606–612.
Morgan G . Granular cell myoblastoma of the orbit. Report of a case. Arch Ophthalmol 1976; 94 (12): 2135–2142.
Muller W, Dahmen HG . Granular cell tumor of the optic nerve. Albrecht Von Graefes Arch Klin Exp Ophthalmol 1978; 207 (3): 181–188.
Drummond JW, Hall DL, Steen WJ, Maxey SA . Granular cell tumor (myoblastoma) of the orbit. Arch Ophthalmol 1979; 97 (8): 1492–1494.
Goldstein BG, Font RL, Alper MG . Granular cell tumor of the orbit: a case report including electron microscopic observations. Ann Ophthalmol 1982; 14 3: 231–232 236-238.
Karcioglu ZA, Hemphill GL, Wool BM . Granular cell tumor of the orbit: case report and review of the literature. Ophthalmic Surg 1983; 14 (2): 125–129.
Moriarty P, Garner A, Wright JE . Case report of granular cell myoblastoma arising within the medial rectus muscle. Br J Ophthalmol 1983; 67 (1): 17–22.
Dolman PJ, Rootman J, Dolman CL . Infiltrating orbital granular cell tumour: a case report and literature review. Br J Ophthalmol 1987; 71 (1): 47–53.
Jaeger MJ, Green WR, Miller NR, Harris GJ . Granular cell tumor of the orbit and ocular adnexae. Surv Ophthalmol 1987; 31 (6): 417–423.
Moseley I . Granular cell tumour of the orbit: radiological findings. Neuroradiology 1991; 33 (5): 399–402.
McNab AA, Daniel SE . Granular cell tumours of the orbit. Aust N Z J Ophthalmol 1991; 19 (1): 21–27.
Rodriguez-Ares T, Varela-Duran J, Sanchez-Salorio M, Varela-Nunez R, Capeans-Tome C, Urdiales-Viedma M . Granular cell tumor of the eye (myoblastoma): ultrastructural and immunohistochemical studies. Eur J Ophthalmol 1993; 3 (1): 47–52.
Allaire GS, Laflamme P, Bourgouin P . Granular cell tumour of the orbit. Can J Ophthalmol 1995; 30 (3): 151–153.
Charles NC, Fox DM, Glasberg SS, Sawicki J . Epibulbar granular cell tumor. Report of a case and review of the literature. Ophthalmology 1997; 104 (9): 1454–1456.
Callejo SA, Kronish JW, Decker SJ, Cohen GR, Rosa RJ . Malignant granular cell tumor metastatic to the orbit. Ophthalmology 2000; 107 (3): 550–554.
Sabet SJ, Tarbet KJ, Lemke BN, Smith ME, Albert DM . Granular cell tumor of the lacrimal sac and nasolacrimal duct: no invasive behavior with incomplete resection. Ophthalmology 2000; 107 (11): 1992–1994.
Ahdoot M, Rodgers IR . Granular cell tumor of the orbit: magnetic resonance imaging characteristics. Ophthal Plast Reconstr Surg 2005; 21 (5): 395–397.
Golio DI, Prabhu S, Hauck EF, Esmaeli B . Surgical resection of locally advanced granular cell tumor of the orbit. J Craniofac Surg 2006; 17 (3): 594–598.
Ayres B, Miller NR, Eberhart CG, Dibernardo CW . Ultrasound features of orbital granular cell tumor. Ophthal Plast Reconstr Surg 2009; 25 (4): 320–322.
von Holstein SL, Ostergaard J, Daugaard S, Toft PB, Heegaard S . Granular cell tumour of the lacrimal gland. Acta Ophthalmol 2009; 87 (8): 926–927.
Poyraz CE, Kiratli H, Soylemezoglu F . Granular cell tumor of the inferior rectus muscle. Korean J Ophthalmol 2009; 23 (1): 43–45.
Sadeghi TA, Asadi Amoli F, Tayari N . Granular cell tumor of ocular adnexa; report of three cases and review of literature. Iran J Ophthalmol 2010; 22 (1): 52–55.
Guerriero S, Giancipoli G, Sborgia A, Fiore MG, Rossi R, Piscitelli D . Orbital granular cell tumor in a patient with Churg Strauss syndrome: the importance of biopsy. Orbit 2011; 30 (1): 30–33.
Ribeiro SF, Chahud F, Cruz AA . Oculomotor disturbances due to granular cell tumor. Ophthal Plast Reconstr Surg 2012; 28 (1): e23–e27.
Fernandes BF, Belfort NR, Odashiro AN, Pereira PR, Burnier JM . Clinical and histopathological features of orbital granular cell tumor: case report. Arq Bras Oftalmol 2012; 75 (2): 137–139.
Salour H, Tavakoli M, Karimi S, Rezaei KM, Faghihi M . Granular cell tumor of the orbit. J Ophthalmic Vis Res 2013; 8 (4): 376–379.
Fisher ER, Wechsler H . Granular cell myoblastoma—a misnomer. Electron microscopic and histochemical evidence concerning its Schwann cell derivation and nature (granular cell schwannoma). Cancer 1962; 15: 936–954.
Aparicio SR, Lumsden CE . Light- and electron-microscope studies on the granular cell myoblastoma of the tongue. J Pathol 1969; 97 (2): 339–355.
Pearse AG . The histogenesis of granular-cell myoblastoma (granular-cell perineural fibroblastoma). J Pathol Bacteriol 1950; 62 (3): 351–362.
Azzopardi JG . Histogenesis of the granular-cell myoblastoma. J Pathol Bacteriol 1956; 71 (1): 85–94.
Garancis JC, Komorowski RA, Kuzma JF . Granular cell myoblastoma. Cancer 1970; 25 (3): 542–550.
Lee D, Suh YL, Nam DH . Cerebral granular cell tumor. Neuropathology 2008; 28 (4): 417–421.
Ordonez NG, Mackay B . Granular cell tumor: a review of the pathology and histogenesis. Ultrastruct Pathol 1999; 23 (4): 207–222.
Chaudhry IH, Calonje E . Dermal non-neural granular cell tumour (so-called primitive polypoid granular cell tumour): a distinctive entity further delineated in a clinicopathological study of 11 cases. Histopathology 2005; 47 (2): 179–185.
Lazar AJ, Fletcher CD . Primitive nonneural granular cell tumors of skin: clinicopathologic analysis of 13 cases. Am J Surg Pathol 2005; 29 (7): 927–934.
Kuhn A, Mahrle G, Steigleder GK . Benign and malignant granular cell tumors. An immunohistochemical classification of tumor cells. Z Hautkr 1987; 62 (12): 952–958.
LeBoit PE, Barr RJ, Burall S, Metcalf JS, Yen TS, Wick MR . Primitive polypoid granular-cell tumor and other cutaneous granular-cell neoplasms of apparent nonneural origin. Am J Surg Pathol 1991; 15 (1): 48–58.
Newton P, Schenker M, Wadehra V, Husain A . A case of metastatic non-neural granular cell tumor in a 13 year old girl. J Cutan Pathol 2014; 41 (6): 536–538.
Gamboa LG . Malignant granular-cell myoblastoma. AMA Arch Pathol 1955; 60 (6): 663–668.
Fanburg-Smith JC, Meis-Kindblom JM, Fante R, Kindblom LG . Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol 1998; 22 (7): 779–794.
Conley AP, Koplin S, Caracciollo JT, Reed DR, Webber NP, Attia S . Dramatic response to pazopanib in a patient with metastatic malignant granular cell tumor. J Clin Oncol 2014; 32 (32): e107–e110.
Acknowledgements
This study was sponsored by grants from the National Nature Science Foundation Project of China (81100656/H1203) and the Shanghai Municipal Natural Science Foundation (12ZR1404900). Provenance and peer review: Not commissioned; externally peer-reviewed.
Author contributions
JQ was involved in design and conduct of study; X-FL, Y-FY and RZ were involved in collection and management of the data; JQ, Y-FY and RZ were involved in provision of patients and resources; X-FL, JQ and Y-WB were involved in analysis and interpretation of the data; X-FL and JQ were involved in the preparation of manuscript; X-FL, JQ, Y-FY, Y-WB and RZ were involved in review and approval of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Li, XF., Qian, J., Yuan, YF. et al. Orbital granular cell tumours: clinical and pathologic characteristics of six cases and literature review. Eye 30, 529–537 (2016). https://doi.org/10.1038/eye.2015.268
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2015.268
This article is cited by
-
Retinal granular cell tumor: a case report
BMC Ophthalmology (2021)
