
Abstract
Purpose
The purpose of this study is to describe the phenotype of a family with de novo mutation in the GUCY2D.
Materials and methods
Five subjects, including two monozygotic twins, underwent ophthalmic clinical examination while some had autofluorescence imaging (AF) and optical coherence tomography (OCT). Symptomatic individuals underwent electrophysiological testing. The youngest subject (21 years) was also evaluated psychophysically. DNA obtained from the individuals was screened for mutations in GUCY2D. Microsatellite markers were used to determine the haplotype of 17p surrounding the GUCY2D gene.
Results
The youngest subject had 6/18 visual acuity, an annulus of hyper-autofluorescence in the perifoveal region, and a subfoveal absence of outer segments on OCT. In the older individuals, severe thinning of inner retina and a patchy loss of photoreceptors and retinal pigment epithelium were observed in the perifoveal region. All three showed generalised cone system dysfunction with preserved rod function on electrophysiology. Psychophysical evaluation was consistent with poor cone function. Screening of the GUCY2D gene revealed the mutation p.R838H in all the affected individuals and was absent in the asymptomatic patients. Haplotyping showed that the mutation originated from the unaffected mother.
Conclusions
Autosomal dominant cone dystrophy due to GUCY2D can occur without a history in the antecedents due to a de novo mutation. This is important to consider in any simplex case with a similar phenotype. The phenotype description of this disorder is expanded with detailed description of the OCT findings. This paper describes the concordance of the phenotypic findings in the monozygotic twins.
Similar content being viewed by others

Introduction
The cone dystrophies and cone–rod dystrophies (CORD) are a heterogeneous group of progressive genetically determined retinal disorders, which may be inherited as an autosomal dominant, autosomal recessive, or X-linked trait.1 They are characterised clinically by a loss of visual acuity, abnormal colour vision, photophobia, and visual field loss. Many will develop macular atrophy. Electroretinography demonstrates generalised cone system dysfunction with either no or mild rod system involvement.2
The gene ‘Guanylate cyclase 2D, membrane (retina-specific)’ (GUCY2D, MIM ID*600179) accounts for up to 35% of patients with autosomal dominant cone dystrophy or CORD.3, 4, 5 It was also the first gene to be implicated in Leber congenital amaurosis, a major cause of blindness in children.6 The protein translated by GUCY2D, retinal guanylate cyclase 1, is expressed in both rod and cone photoreceptors.7 Two mutation sites in GUCY2D, the codons 575 and 838, have been reported to cause autosomal dominant cone dystrophy or CORD.5, 8 Recently, the mutation p.V933A was described in GUCY2D associated with the phenotype of autosomal dominant central areolar choroidal dystrophy, a condition similar to CORD.9 The importance of identifying this condition is paramount as, using a gene therapy technique, a partial restoration of visual function has been demonstrated in a GUCY2D knockout mouse model increasing the likelihood of eventual treatment in humans.10
The present report describes a family with autosomal dominant cone dystrophy due to a de novo mutation in the codon 838 in the GUCY2D gene.
Materials and methods
The study adhered to the tenets of the Declaration of Helsinki. The family were presented to the inherited eye disease clinic at the Moorfields Eye Hospital NHS Foundation Trust, London, UK. All subjects provided informed consent to participate in the study.
Clinical investigations
All participants underwent routine clinical examination, including best-corrected visual acuity, slit-lamp examination, applanation tonometry, and fundoscopy. Colour fundus photographs were obtained using a Topcon TRC 50IA retinal camera (Topcon Corporation, Tokyo, Japan). Fundus autofluorescence imaging (AF) and optical coherence tomography (OCT) were performed using a Spectralis Spectral-domain OCT (Heidelberg Engineering, Heidelberg, Germany). Electrophysiological assessment was performed in the three affected patients using techniques that incorporated the ISCEV standards.11, 12 Additional On–Off ERGs were performed using an orange stimulus (560 cd/m2, duration 200 ms) superimposed on a constant green background (150 cd/m2). S-cone ERGs were performed using a blue stimulus (445 nm, duration 5 ms, 80 cd/m2) superimposed on a constant orange background (620 nm, 560 cd/m2).13
Psychophysics
The psychophysical measurements were made using a Maxwellian-view optical system. Wavelengths were selected with interference filters with full-width at half-maximum bandwidths of between 7 and 11 nm (Ealing, South Natick, MA, USA or Oriel, Stratford, CT, USA). The radiance of each beam could be controlled by the insertion of fixed neutral density filters (Oriel) or by the rotation of circular, variable neutral density filters (Rolyn Optics, Covina, CA, USA). Sinusoidal (flicker) modulation was produced by the pulse-width modulation of fast, liquid crystal light shutters (Displaytech, Longmont, CO, USA) at a carrier frequency of 400 Hz (which is much too fast to be visually resolved). In these experiments, the targets were flickered at 92% modulation, the maximum modulation that could be produced with these shutters. The position of the observer’s head was maintained by a dental wax impression. The system is described in more detail elsewhere.14, 15
A flickering target of 4° of visual angle in diameter was presented in the centre of a 9° diameter background field in all experiments. Fixation was central. Each datum point for the individual measurements represents the average of at least three settings. For the L-cone critical flicker fusion measurements (c.f.f.), the wavelength of the target was fixed at 650 nm and its radiance varied from 6.5 to 11.0 log10 quanta/s/degree2 in steps of about 0.3 log10 units. The target was superimposed upon the centre of a 481 nm background that delivered 8.29 log10 quanta/s/degree2 at the cornea. This background mainly served to suppress the rods, but also selectively desensitised the M-cones at lower target radiances. These conditions isolate the L-cone response in the normal observer over most of the 650 nm intensity range, but at high intensities the M-cones may also contribute to flicker detection. Observers adjusted the flicker frequency to find the frequency at which the flicker just disappeared (the so called critical flicker fusion).
Molecular genetics
The GUCY2D gene was screened for disease-causing mutations after extracting the genomic DNA from peripheral blood leukocytes (Nucleon BACC2 kit; GE Healthcare, Little Chalfont, UK). Primers were designed to amplify the coding region and the intron–exon boundaries of 20 published exons (NM_000180.3). PCRs were performed using the standard protocols, and the PCR products were analysed and purified. Bi-directional Sanger sequencing of the PCR products was performed using BigDye v3.1 (BigDye Terminator sequencing Cycle Sequencing Ready Reaction kit version 1.1) on a 3730 genetic analyzer (Applied Biosystems (ABI), Warrington, UK). DNAStar sequencing analysis software (Lasergene v8.0, DNA Star Inc., Madison, WI, USA) was used to compare electropherograms with those from control samples.
Four microsatellite markers (D17S1828, D17S1876, D17S1791, and D17S799) were used to determine the haplotypes of a 9.4-Mb region in chromosome 17p surrounding the GUCY2D gene. A multiplex PCR was performed according to the manufacturer’s instructions (Applied Biosystems (ABI)), and the pooled amplified products were genotyped using a 3730 genetic analyzer (Applied Biosystems (ABI)). The electropherograms were analysed using the GeneMapper Software version 4.0 (Applied Biosystems (ABI)).
We certify that all applicable institutional and governmental regulations concerning the ethical use of human volunteers were followed during this research.
Results
Clinical phenotype
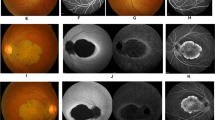
The clinical data on the subjects are summarised in Table 1 and illustrated in Figure 1.

Data from affected subjects IV:1 (a), III:2 (b), III:3 (c), and unaffected subject II:2 (d). The fundus photographs show central macular atrophy in III:2 and III:3 (44-year olds) and a normal macula in subjects (IV:, aged 21 years) and II:2 (unaffected; 65 years old). The autofluorescence images show a hyper-autofluorescent ring surrounding the central macular atrophy in all the affected subjects, worse with age. The OCT reveals absence of outer retinal layers in the central macula in III:2 and III:3 and absence of outer segments in the young IV:1.
The earliest symptom experienced by the affected subjects was reduced central vision. The youngest subject (21-year old) noticed deterioration of her visual acuity at 11 years of age. There was a discordance in onset of symptoms in the monozygotic twins (44-year old) with III:3 noticing problems a decade earlier than III:2. Later in the disorder, photoaversion and a difficulty in colour vision developed. No subject reported nyctalopia.
The best-corrected visual acuity of subject IV:1 was 6/18 while vision was less than 6/60 in subjects III:2 and III:3. The reduction of vision in the right eye of subject II:1 was from an advanced cataract. He had undergone retinal reattachment surgery in his left eye. Visual acuity was normal in subject II:2. Colour vision (Ishihara) was very poor in subjects III:2, III:3, and IV:1.
Fundus examination of IV:1 showed mild pigmentary changes in the macula. The fundi of the monozygotic twins showed symmetrical well-demarcated perifoveal retinal thinning. The retina of the right eye of II:1 was difficult to examine due to dense cataract. The left eye showed peripapillary atrophy and an abnormal foveal reflex. Subject II:2 showed no abnormality on fundus examination.
Fundus AF of IV:1 showed a small area of mild hypo-autofluorescence in the perifoveal region surrounded by a ring of hyper-autofluorescence. There was progressive decrease in the foveal autofluorescence with age (not shown). Subjects III:2 and III:3 both show a larger central area of speckled hypo-autofluorescence, reflecting patchy atrophic changes in the retinal pigment epithelium (RPE), surrounded by a ring of increased autofluorescence. Subject II:2 had normal AF. FAF could not be performed reliably in II:1 due to cataracts.
OCT was performed in subject IV:1, aged 21 years, and showed abrupt disruption of the inner–outer segment junction layer in the subfoveal photoreceptors. In her mother and aunt (III:2 and III:3), there is thinning of the inner retina with irregular disruption of photoreceptor–RPE complex. The junction between normal and abnormal outer retina corresponded to the hyper-autofluorescent ring referred to above. The OCT was within normal limits in II:2 and was not performed in II:1.
In one of the monozygotic twins III:2, the areas of speckled hypo-autofluorescence were 6.99 and 6.33 mm2 in the right and left eye, respectively, while in case of her twin sister the areas were 6.89 and 6.83 mm2 in the right and left eye, respectively. The b-wave amplitudes of photopic cone ERG in III:2 was 65% on the right and 75% on the left eye in comparison to the lower limit of normal while the amplitudes were 68 and 72% of the lower limit of normal on the right and left eye, respectively, in her monozygotic twin III:3.
Functional phenotype
Dark-adapted full-field ERGs (DA 0.01, DA11.0) were normal in III:2, III:3, and IV:1, in keeping with preserved rod system function (Figure 2a). Photopic 30 Hz flicker ERGs (LA 3.0 30 Hz) showed mild delay (1–4 ms above the upper limit of normal) and amplitude reduction. Single flash cone ERGs (LA 3.0 2 Hz) showed borderline delay with a subnormal b-wave:a-wave ratio in all subjects, suggesting generalised system dysfunction at an inner retinal level. This was confirmed by On–Off ERGs, which showed electronegative On-responses (reduced b-waves); Off- response d-waves were of borderline timing and, although of normal amplitude, showed an abnormal positive ‘plateau’ following the d-wave. S-cone ERGs were markedly subnormal. Pattern ERGs were undetectable in all the three cases in keeping with severe macular involvement (data not shown).

(a) Full-field electroretinograms (ERGs) from one eye of subjects IV:1 (row A), III:2 (row B) and III:3 (row C) and representative normal traces (bottom row). Dark-adapted ERGs are normal in all three patients. Cone single flash and flicker ERGs are markedly subnormal with reduction in the b:a ratio of the single flash photopic ERGs (LA 3.0) suggestive of inner retinal dysfunction, confirmed by electronegative On- responses to long-duration stimuli. Broken lines replace blink artifacts. (b) L-cone critical flicker fusion measurements (grey hexagons) for affected female (IV:1) compared with the mean measurements (black squares) for 12 normal subjects. Error bars are ±1 SE of the measurements for IV:1 and±1 SE between subjects for the normal data.
Psychophysical examination of the cone system was performed in the 21-year-old female (IV:1). L-cone c.f.f. was measured as a function of the radiance of a 650 nm target superimposed in the center of a 480 nm background of 8.26 log10 quanta/s/degree2. Figure 2b shows the c.f.f. results for the left eye of the affected female (gray hexagons). For comparison, the mean data for 12 normal observers are also shown (black squares). With increasing target radiance, the normal L-cone c.f.f. function starts to rise just above 6.5 log10 quanta/sec//degree2 and then continues to rise steadily until reaching a plateau at 43 Hz (see also, eg, Hecht and Shlaer16 and Hecht and Verrijp17). The affected female shows a devastating loss of L-cone (and M-cone) mediated sensitivity; the target had to be set to almost 100 times brighter than for normal observers for the affected observer to first see flicker, and the c.f.f. reached only 7 Hz.
Molecular genetics
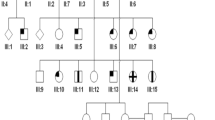
Sanger sequencing of the GUCY2D gene was performed in all the five subjects. Three out of the five (IV:1, III:2, and III:3) carried heterozygous mutation c.2513G>A, p.R838H. The parents (II:1 and II:2) did not carry the mutation. Microsatellite markers surrounding the GUCY2D gene showed that the mutation segregated with the markers derived from the mother (II:2) (Figure 3).

Pedigree of the family of cone dystrophy with p.R838H mutation in GUCY2D. The affected individuals are represented by dark symbols. The electropherograms demonstrating the gene sequence in forward and reverse are demonstrated. The chromosomes of individuals are demonstrated using microsatellite markers and are coded.
Discussion
This study describes the detailed phenotype of patients suffering from autosomal dominant cone dystrophy consequent upon a de novo mutation in GUCY2D, reports the first de novo mutation in the gene, compares the phenotype in the monozygotic twins, and expands our knowledge of the clinical, electrophysiological, and psychophysical phenotypes of the disorder.
Onset of the disease as evidenced by a decrease in the visual acuity was variable between early teens to early twenties. There was relative concordance among the monozygotic twins when the areas of speckled hypofluorescence and the b-wave amplitudes of the photopic cone ERG were compared. However, there was a discordance of onset between the two monozygotic twins of more than a decade although the final visual acuity was similar at 44 years of age. Similar variability in the age on onset has been described in patients with CORD consequent upon GUCY2D mutations.18, 19, 20, 21
All three affected patients in our family had generalised cone dysfunction with severe macular involvement, but unlike the majority of previous cases,18, 19, 20, 21 there was no ERG evidence of rod system dysfunction. The possibility that rod dysfunction will develop later in life cannot be excluded, but there is no evidence of scotopic ERG reduction with increasing age in this small cohort. An electronegative dark-adapted ERG (b:a ratio<1.0) has been reported in GUCY2D CORD,18 but in our family, only the light-adapted ERG had a low b:a ratio. The combination of a flicker ERG delay, an electronegative On- response and the unusual shape of the Off- ERG waveform distinguishes these cases from those with autosomal dominant cone dystrophy due to GUCA1A mutation, often associated with a reduced cone ERG of normal timing with subnormal On- and Off- ERG components.20 The latter study speculated that these abnormalities could result from impaired transduction kinetics, but there is also evidence of weak RetGC expression in the outer plexiform layer22 and histological evidence of synaptic disruption in cases of CORD.23 Generalised cone dysfunction was accompanied by perifoveal photoreceptor–RPE disruption in the monozygotic twins. This structural loss of the central cones may result from the high cone density.
In the phototransduction cascade, exposure to a photon results in a decrease in the photoreceptor intracellular Ca2+ concentration. This is because light leads to sequential isomerisation of rhodopsin and activation of transducin and phosphodiesterase (PDE). PDE hydrolyses cGMP closing cGMP-gated cation channels ceasing the calcium influx. RetGC-1, the protein translated by GUCY2D is involved in the regeneration of cGMP in the photoreceptors. This is achieved in response to lowering of Ca2+ concentration under the influence of guanylate cyclase activating proteins (GCAP). The mutation in this report, p.R838H, is believed to cause a gain of function increasing the affinity of RetGC-1 for GCAP even in high Ca2+ concentrations.24 The cone photoreceptor death in this disorder is believed to be caused by the high cGMP concentration keeping cGMP-gated cation channels open, resulting in increased Ca2+ concentration in the cell.25 Decreasing the cGMP concentration may be therapeutic for these individuals. This can potentially be achieved by increasing photopic exposure of the photoreceptors resulting in stimulation of PDE26 or can be directly achieved by administration of PDE agonists. Interestingly, PDE6C recessive mutations abrogating the PDE function have been described to cause a phenotype of early-onset cone photoreceptor dystrophy.27
Haplotype analysis of our family confirmed a de novo mutation in GUCY2D transmitted from the unaffected mother. However, de novo mutations more commonly derive from the paternal germline.28 Ascertainment of a family showing a de novo mutation in any inherited disease affects the counselling advice given to a person presenting with the disorder. The diagnosis of the disorder in an individual without a family history would usually suggest a recessive inheritance. The recurrence risk to future siblings in such a pedigree is extremely low but might occur if the de novo mutation involved the parent’s germ-cells, the so called germline mosaicism. However, to date, there have been no reports of this occurring for genes involved in retinal degeneration. Given the findings in this family, the possibility of a de novo mutation causing a dominant allele in GUCY2D needs to be considered as this would dramatically change the risks of recurrence in children from a population risk to 50%. The prevalence of families presenting this way is not known but will become clearer with the implementation of high-throughput sequencing. Alternatively, in cone dystrophy, specific tests for dominant alleles in GUCY2D and/or GUCA1A should be considered when seeking a molecular diagnosis even in the absence of a family history.
To conclude, this report expands and refines the phenotypic description of patients with the cone dystrophy associated with mutations in GUCY2D gene. The existence of a de novo germline mutation herein described should alert the physician to its possibility in a family with cone dystrophy and has implications for counselling.

References
RetNet. . Available from http://www.sph.uth.tmc.edu/RetNet/.
Michaelides M, Hardcastle AJ, Hunt DM, Moore AT . Progressive cone and cone-rod dystrophies: phenotypes and underlying molecular genetic basis. Surv Ophthalmol 2006; 51 (3): 232–258.
Kitiratschky VB, Nagy D, Zabel T, Zrenner E, Wissinger B, Kohl S et al. Cone and cone-rod dystrophy segregating in the same pedigree due to the same novel CRX gene mutation. Br J Ophthalmol. [Research Support, Non-U.S. Gov't] 2008; 92 (8): 1086–1091.
Payne AM, Morris AG, Downes SM, Johnson S, Bird AC, Moore AT et al. Clustering and frequency of mutations in the retinal guanylate cyclase (GUCY2D) gene in patients with dominant cone-rod dystrophies. J Med Genet 2001; 38 (9): 611–614.
Hunt DM, Buch P, Michaelides M . Guanylate cyclases and associated activator proteins in retinal disease. Mol Cell Biochem 2010; 334 (1-2): 157–168.
Perrault I, Rozet JM, Calvas P, Gerber S, Camuzat A, Dollfus H et al. Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat Genet 1996; 14 (4): 461–464.
Yang RB, Foster DC, Garbers DL, Fulle HJ . Two membrane forms of guanylyl cyclase found in the eye. Proc Natl Acad Sci US A 1995; 92 (2): 602–606.
Small KW, Silva-Garcia R, Udar N, Nguyen EV, Heckenlively JR . New mutation, P575L, in the GUCY2D gene in a family with autosomal dominant progressive cone degeneration. Arch Ophthalmol 2008; 126 (3): 397–403.
Hughes AE, Meng W, Lotery AJ, Bradley DT . A novel GUCY2D mutation, V933A, causes central areolar choroidal dystrophy. Invest Ophthalmol Vis Sci. [Research Support, Non-U.S. Gov't] 2012; 53 (8): 4748–4753.
Boye SE, Boye SL, Pang J, Ryals R, Everhart D, Umino Y et al. Functional and behavioral restoration of vision by gene therapy in the guanylate cyclase-1 (GC1) knockout mouse. PLoS One 2010; 5 (6): e11306.
Holder GE, Brigell MG, Hawlina M, Meigen T, Vaegan, Bach M . ISCEV standard for clinical pattern electroretinography—2007 update. Doc Ophthalmol 2007; 114 (3): 111–116.
Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M . ISCEV standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol 2009; 118 (1): 69–77.
Arden G, Wolf J, Berninger T, Hogg CR, Tzekov R, Holder GE . S-cone ERGs elicited by a simple technique in normals and in tritanopes. Vis Res 1999; 39 (3): 641–650.
Stockman A, Sharpe LT, Tufail A, Kell PD, Ripamonti C, Jeffery G . The effect of sildenafil citrate (Viagra) on visual sensitivity. J Vis 2007; 7 (8): 4.
Stockman A, Smithson HE, Webster AR, Holder GE, Rana NA, Ripamonti C et al. The loss of the PDE6 deactivating enzyme, RGS9, results in precocious light adaptation at low light levels. J Vis 2008; 8 (1): 10.1–10.10.
Hecht S, Shlaer S . Intermittent stimulation by light: V. The relation between intensity and critical frequency for different parts of the spectrum. J Gen Physiol 1936; 19 (6): 965–977.
Hecht S, Verrijp CD . Intermittent stimulation by light: IV. A theoretical interpretation of the quantitative data of flicker. J Gen Physiol 1933; 17 (2): 269–282.
Gregory-Evans K, Kelsell RE, Gregory-Evans CY, Downes SM, Fitzke FW, Holder GE et al. Autosomal dominant cone-rod retinal dystrophy (CORD6) from heterozygous mutation of GUCY2D, which encodes retinal guanylate cyclase. Ophthalmology 2000; 107 (1): 55–61.
Smith M, Whittock N, Searle A, Croft M, Brewer C, Cole M . Phenotype of autosomal dominant cone-rod dystrophy due to the R838C mutation of the GUCY2D gene encoding retinal guanylate cyclase-1. Eye 2007; 21 (9): 1220–1225.
Downes SM, Payne AM, Kelsell RE, Fitzke FW, Holder GE, Hunt DM et al. Autosomal dominant cone-rod dystrophy with mutations in the guanylate cyclase 2D gene encoding retinal guanylate cyclase-1. Arch Ophthalmol 2001; 119 (11): 1667–1673.
Ito S, Nakamura M, Ohnishi Y, Miyake Y . Autosomal dominant cone-rod dystrophy with R838H and R838C mutations in the GUCY2D gene in Japanese patients. Jpn J Ophthalmol. [Case Reports Research Support, Non-U.S. Gov't] 2004; 48 (3): 228–235.
Liu X, Seno K, Nishizawa Y, Hayashi F, Yamazaki A, Matsumoto H et al. Ultrastructural localization of retinal guanylate cyclase in human and monkey retinas. Experimental eye research. [Research Support, Non-U.S. Gov't] 1994; 59 (6): 761–768.
Gregory-Evans K, Fariss RN, Possin DE, Gregory-Evans CY, Milam AH . Abnormal cone synapses in human cone-rod dystrophy. Ophthalmology. [Case Reports Comparative Study Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S.] 1998; 105 (12): 2306–2312.
Wilkie SE, Newbold RJ, Deery E, Walker CE, Stinton I, Ramamurthy V et al. Functional characterization of missense mutations at codon 838 in retinal guanylate cyclase correlates with disease severity in patients with autosomal dominant cone-rod dystrophy. Hum Mol Genet 2000; 9 (20): 3065–3073.
Tucker CL, Woodcock SC, Kelsell RE, Ramamurthy V, Hunt DM, Hurley JB . Biochemical analysis of a dimerization domain mutation in RetGC-1 associated with dominant cone-rod dystrophy. Proc Natl Acad Sci USA 1999; 96 (16): 9039–9044.
Liu X, Pawlyk BS, Adamian M, Olshevskaya EV, Dizhoor AM, Makino CL et al. Increased light exposure alleviates one form of photoreceptor degeneration marked by elevated calcium in the dark. PLoS One. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t] 2009; 4 (12): e8438.
Thiadens AA, den Hollander AI, Roosing S, Nabuurs SB, Zekveld-Vroon RC, Collin RW et al. Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am J Hum Genet 2009; 85 (2): 240–247.
Ellegren H . Characteristics, causes and evolutionary consequences of male-biased mutation. Proc Biol Sci 2007; 274 (1606): 1–10.
Acknowledgements
This research was supported by grants from The Foundation Fighting Blindness, Fight for Sight, National Institute for Health Research Biomedical Research Centre for Ophthalmology, BBSRC, and the British Retinitis Pigmentosa Society. We would like to thank the patients who participated in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Mukherjee, R., Robson, A., Holder, G. et al. A detailed phenotypic description of autosomal dominant cone dystrophy due to a de novo mutation in the GUCY2D gene. Eye 28, 481–487 (2014). https://doi.org/10.1038/eye.2014.7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2014.7
