
Abstract
Purpose
To describe the spectrum of phenotypic characteristics of BEST1-related autosomal dominant vitreoretinochoroidopathy (ADVIRC) in a family with p.V86M mutation.
Methods
A retrospective review of the clinical, psychophysical, and electrophysiological phenotypes of six subjects with ADVIRC. Five family members were sequenced for mutations in the BEST1gene.
Results
A heterozygous change, p.V86M (c.256G>A), was identified in the BEST1gene in the three affected subjects tested, and was shown to segregate with the disease phenotype. The distance visual acuity ranged from ⩾20/25 to absent perception of light. Clinical features observed included angle closure glaucoma (n=2), microcornea with shallow anterior chamber (n=1), iris dysgenesis (n=2), cataracts (n=4), classical peripheral concentric band of retinal hyperpigmentation (n=5), and optic nerve dysplasia (n=1). Full-field electroretinogram response amplitudes ranged from low normal (two cases; 27 and 32 years) to non-recordable (two cases; 42 and 63 years). Goldmann fields were normal in two (27 and 28 years) but were abnormal in two older subjects. Optical coherence tomography showed macular thinning in the proband, whereas his affected daughter had normal macular thickness. Electro-oculography showed borderline Arden's ratio (1.50) in the lone case tested (27 years).
Conclusion
ADVIRC is a slowly progressive vitreoretinal degeneration that demonstrates marked intra-familial phenotypic variability. Optic nerve dysplasia and iris dysgenesis are novel observations that extend the ocular phenotype of ADVIRC.
Similar content being viewed by others
Introduction
Autosomal dominant vitreoretinochoroidopathy (ADVIRC) is classically characterized by a circumferential band of peripheral retinal hyperpigmentation with a well-defined posterior demarcation1 and midperipheral chorioretinal atrophy. Associated ocular findings1, 2, 3, 4 include microcornea, microphthalmos, angle closure glaucoma, cataract, fibrillary vitreous condensations, disc gliosis, macular oedema, intraretinal white deposits, and preretinal neovascularization.
Mutations causing ADVIRC were first identified in BEST1 gene in five pedigrees,5 which also included families previously reported in literature.2, 3, 6 BEST1 encodes a 585 amino acid transmembrane protein bestrophin-1, located in the basolateral membrane of the retinal pigment epithelium (RPE),7 and was originally associated with Best vitelliform macular dystrophy (BVMD).8
We describe the phenotypic variability of ADVIRC in a family with mutations in BEST1 (p.V86M), and report novel ocular associations including iris dysgenesis and optic nerve dysplasia.
Subjects and methods
The study was approved by the Research Ethics Board of The Hospital for Sick Children. Retrospective chart review of six affected subjects and related family members was performed. Details of ophthalmological evaluation, psychophysical testing and full-field electroretinography (ERG) were obtained. Goldmann visual field (GVF) results were available on four subjects. Stratus optical coherence tomography (OCT) was performed on two. Sensory electro-oculogram (EOG), fundus autofluorescence (FAF), and fundus fluorescein angiogram (FFA) were only available on one each. Direct sequencing of the BEST1 gene was performed by the Carver Laboratory on five family members including three affected individuals.
Results
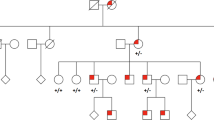
Family members had been assessed at The Hospital for Sick Children at different times over the last 19 years. Mutation screening of the BEST1 coding region identified a previously reported heterozygous change, p.V86M (c.256G>A),5 in all three tested patients, and the mutation was shown to segregate with the disease phenotype. Figure 1 is a four generation pedigree demonstrating the co-segregation and the autosomal dominant inheritance pattern.

Pedigree of the four generation family studied demonstrates autosomal dominant inheritance pattern. Solid symbols indicate affected status and symbols with a slash indicate deceased individuals. The heterozygous change p.V86M was noted in three affected and it segregated with the disease.
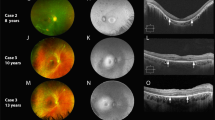
The detailed phenotypic characteristics of all six cases are given in Table 1. The best corrected visual acuity ranged from ⩾20/25 (four cases; age range 27–61 years) to absent perception of light (case II1; 63 years). Colour vision was normal in all four tested. Angle closure glaucoma was documented in two (cases II1 and II2), whereas microcornea with shallow anterior chamber was noted in one (case IV2). Iris dysgenesis with abnormal pupillary ruff (Figure 2a and f) was observed in two (cases III2 and IV2). All except case IV2 either had cataracts (n=4; presenile in cases III6 and III5; Figure 2i) or had a history of cataract surgery (case III2). The classical concentric band of retinal hyperpigmentation (Figure 2d and j) was noted in all except one (case II1), but the posterior extent of retinal pigmentation was variable. Case II1 was blind with dense cataracts at diagnosis that precluded retinal evaluation, but had angle closure glaucoma, non-recordable ERG, and she had the V86M mutation. Bilateral optic nerve dysplasia was noted in one (case IV2; Figure 2b).

(a–e) Phenotypic characteristics of case IV2 (a) Anterior segment photo of right eye demonstrating microcornea and iris dysgenesis. Note the absence of krypts and abnormal pupillary ruff. (b) Fundus photograph of the left eye demonstrating optic nerve dysplasia and otherwise normal posterior pole. The inset photograph is a magnified view of the dysplastic disc. (c) Fundus autofluorescence image showing normal autofluorescence pattern in the posterior pole in the left eye. (d) Fundus photograph demonstrating characteristic concentric band of hyperpigmentation in the extreme periphery of one quadrant. (e) Optical coherence tomography (OCT) of the right eye showing normal central retinal thickness. (f–h) Phenotypic characteristics of Case III 2. (f) Anterior segment photograph showing normal corneal diameter, iris dysgenesis and pseudophakia. (g) Fundus photograph of the right eye posterior pole at 58 years showing pale optic disc, markedly attenuated vessels, background pigmentary changes and advanced chorioretinal atrophy. (h) OCT of the left eye at 58 yrs demonstrating significant central retinal thinning. (i–k) Phenotypic characteristics of Case III 5. (i) Anterior segment photograph demonstrating posterior subcapsular cataract (32 years). (j) Fundus photograph of the periphery at the same visit showing characteristic concentric band of retinal hyperpigmentation with a posterior demarcation border. (k, l) Fundus fluorescein angiography showed blocked fluorescence at the region of hyperpigmentation, hyperfluorescence along the posterior border of the lesion suggesting retinal atrophy and the severe atrophy of the underlying choroidal vessels.
GVF was normal in younger individuals (cases III5 and IV2), but the progression was variable (Table 1). The ERG amplitudes ranged from low normal to non-recordable (Table 1). However, three of the four cases with recordable ERG (except case III6) had normal implicit times. The EOG showed borderline Arden's ratio (1.50) in the lone subject tested (case IV2; 27 years). Case IV2 had normal autofluorescence on FAF (Figure 2c), and also demonstrated normal macular thickness on OCT (Figure 2e). However, her father (case III2 at age 58) demonstrated macular thinning on OCT (Figure 2h). The FFA (case III5) showed marked RPE atrophy along the demarcation border with underlying atrophy of choroidal vessels (Figure 2k and l).
The proband, case III2 (42 years at presentation) demonstrated a progressive phenotype over 16 years (Table 1). The central vision in the right eye demonstrated rapid worsening to light perception by 44 years of age. The left eye kept residual tunnel vision for another 10 years. Over the period, he progressed to have severe retinal vascular thinning, chorioretinal atrophy, and optic disc pallor (Figure 2g).
Discussion
ADVIRC described as a vitreoretinochoroidopathy,1 is also associated with developmental ocular anomalies including microcornea,2, 3 angle closure glaucoma,2, 3 and cataract.2, 4 We noted developmental anomalies, such as iris dysgenesis and optic nerve dysplasia, as novel associations of ADVIRC. BEST1 is proposed to have a role in normal ocular development.5 Ex vivo studies reveal that, all BEST1 mutations known to cause ADVIRC (including p.V86M mutation5) result in aberrant mRNA splicing, producing bestrophin isoforms with internally deleted,5 or duplicated9 exons. This might account for the distinct ocular phenotype and high incidence of developmental anomalies in ADVIRC.
There is marked intra-familial phenotypic variability with regards to disease severity and associated developmental ocular abnormalities, such as microcornea, presenile cataracts, angle closure glaucoma, iris dysgenesis, and optic nerve dysplasia. The concentric band of retinal hyperpigmentation is a constant feature regardless of age. The disease appears to be slowly progressive with regards to vision loss, field loss, and retinal dysfunction. The visual fields are intact early in the course and central fields appear to be affected late as previously reported,3 but pattern of loss and rate of progression are variable. The ERG amplitudes varied between individuals but tended to worsen with increasing age. The preservation of normal implicit times in subjects with measurable ERG suggests absence of gross outer and inner retinal dysfunction in the residual retina. These results are in keeping with earlier studies3 and suggest slow disease progression. Bestrophin-1 is currently thought to modulate light peak depolarization through its interaction with voltage-dependant Ca2+ channels.10, 11 Although the p.V86M change in BEST-1 has always been associated with abnormal EOG,3, 5 the borderline EOG light rise noted in one case probably represents a milder phenotype identified early in the disease course.
In addition to ADVIRC, mutations in BEST1 gene also cause other ocular phenotypes, such as BVMD,12 adult-onset foveomacular vitelliform dystrophy13 (AFVD) and autosomal recessive bestrophinopathy14 (ARB). BVMD is autosomal dominant, AFVD is usually sporadic, and ARB is an autosomal recessive disease. Clinically, BVMD15 and AFVD16 present with vitelliform lesion at the macula that progress through stages over time but usually the central vision is preserved until late in the disease. In comparison with BVMD, AFVD16 is a milder disease with a later onset and slower progression. In contrast, ARB presents early in life with central visual acuity loss, irregular RPE alterations, and whitish sub-retinal deposits,14, 17 and is associated with higher incidence of angle closure glaucoma.14 The sensory EOG is usually severely abnormal in all BEST1-related diseases, except AFVD, which usually demonstrates normal Arden's ratio.16 The ERG is usually normal in BVMD18 and AFVD.16 ARB is associated with reduced amplitude and delayed implicit times of rod and cone responses.14, 17 In ADVIRC, ERG amplitudes may vary between normal and non-detectable, depending on the severity of the phenotype. The vitelliform lesion in both BVMD and AFVD is hyper-reflective on OCT and may be associated with sensory detachment of the macula.19 In ADVIRC, as noted in this report, the macular OCT is usually normal early in the disease and becomes thinned out as disease progresses.
ADVIRC is a slowly progressive distinct bestrophinopathy with marked intra-familial phenotypic variability. Serial evaluations are required to determine the rate of progression of retinal phenotype. The anterior segment developmental anomalies should be identified early and monitored closely as it is associated with significant risk of developing glaucoma.

References
Kaufman SJ, Goldberg MF, Orth DH, Fishman GA, Tessler H, Mizuno K . Autosomal dominant vitreoretinochoroidopathy. Arch ophthalmol 1982; 100: 72–78.
Hermann P . Le syndrome microphtalmie-rétinite pigmentaire-glaucome. Archives d’ophtlamologie et revue general d’ophtalmologie 1958; 18: 12–24.
Lafaut BA, Loeys B, Leroy BP, Spileers W, De Laey JJ, Kestelyn P . Clinical and electrophysiological findings in autosomal dominant vitreoretinochoroidopathy: report of a new pedigree. Graefe's Arch Clin Exp Ophthalmol 2001; 239: 575–582.
Traboulsi EI, Payne JW . Autosomal dominant vitreoretinochoroidopathy. Report of the third family. Arch Ophthalmol 1993; 111: 194–196.
Yardley J, Leroy BP, Hart-Holden N, Lafaut BA, Loeys B, Messiaen LM et al. Mutations of BEST1 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC). Invest Ophthalmol Vis Sci 2004; 45: 3683–3689.
François P, Puech B, Hache JC, Lafinneur Q . Hérédo-dystrophie choriorétinovitréenne, microcornée, glaucome et cataracte. J Fr Ophtalmol 1993; 16: 29–40.
Marmorstein AD, Marmorstein LY, Rayborn M, Wang X, Hollyfield JG, Petrukhin K . Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc Natl Acad Sci USA 2000; 97: 12758–12763.
Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet 1998; 19: 241–247.
Burgess R, MacLaren RE, Davidson AE, Urquhart JE, Holder GE, Robson AG et al. ADVIRC is caused by distinct mutations in BEST1 that alter pre-mRNA splicing. J Med Genet 2009; 46: 620–625.
Marmorstein LY, Wu J, McLaughlin P, Yocom J, Karl MO, Neussert R et al. The light peak of the electroretinogram is dependent on voltage-gated calcium channels and antagonized by bestrophin (Best-1). J Gen Physiol 2006; 127: 577–589.
Zhang Y, Stanton JB, Wu J, Yu K, Hartzell HC, Peachey NS et al. Suppression of Ca2+ signaling in a mouse model of Best disease. Hum Mol Genet 2010; 19: 1108–1118.
Best F . Uber eine hereditare Maculaaffektion. Beitrage zur Vererbungslehre. Zeitschrift fuir Augenheilkuntde 1905; 13: 199–212.
Gass JD . A clinicopathologic study of a peculiar foveomacular dystrophy. Trans Am Ophthalmol Soc 1974; 72: 139–156.
Burgess R, Millar ID, Leroy BP, Urquhart JE, Fearon IM, De Baere E et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am J Hum Genet 2008; 82: 19–31.
Gass JD . Heredodystrophic disorders affecting the pigment epithelium and retina. In: Gass JD (ed). Stereoscopic Atlas of Macular diseases: Diagnosis and Treatment. CV Mosby: St Louis, 1997, pp 304–325.
Renner AB, Tillack H, Kraus H, Kohl S, Wissinger B, Mohr N et al. Morphology and functional characteristics in adult vitelliform macular dystrophy. Retina 2004; 24: 929–939.
Gerth C, Zawadzki RJ, Werner JS, Héon E . Detailed analysis of retinal function and morphology in a patient with autosomal recessive bestrophinopathy (ARB). Doc Ophthalmol 2009; 118: 239–246.
Renner AB, Tillack H, Kraus H, Krämer F, Mohr N, Weber BH et al. Late onset is common in best macular dystrophy associated with VMD2 gene mutations. Ophthalmology 2005; 112: 586–592.
Pianta MJ, Aleman TS, Cideciyan AV, Sunness JS, Li Y, Campochiaro BA et al. In vivo micropathology of Best macular dystrophy with optical coherence tomography. Exp Eye Res 2003; 76: 203–211.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Vincent, A., McAlister, C., VandenHoven, C. et al. BEST1-related autosomal dominant vitreoretinochoroidopathy: a degenerative disease with a range of developmental ocular anomalies. Eye 25, 113–118 (2011). https://doi.org/10.1038/eye.2010.165
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2010.165
Keywords
This article is cited by
-
Long-term changes in autosomal dominant vitreoretinochoroidopathy (ADVIRC)
Graefe's Archive for Clinical and Experimental Ophthalmology (2018)
