
Abstract
Mice deficient in the toll-like receptor (TLR) or the myeloid differentiation factor 88 (MyD88) are resistant to acute liver failure (ALF) with sudden death of hepatocytes. Chalcone derivatives from medicinal plants protect from hepatic damages including ALF, but their mechanisms remain to be clarified. Here, we focused on molecular basis of piperidylmethyloxychalcone (PMOC) in the treatment of TLR/MyD88-associated ALF. C57BL/6J mice were sensitized with D-galactosamine (GalN) and challenged with Escherichia coli lipopolysaccharide (LPS, TLR4 agonist) or oligodeoxynucleotide containing unmethylated CpG motif (CpG ODN, TLR9 agonist) for induction of ALF. Post treatment with PMOC sequentially ameliorated hepatic inflammation, apoptosis of hepatocytes, severe liver injury and shock-mediated death in ALF-induced mice. As a mechanism, PMOC inhibited the catalytic activity of TGF-β-activated kinase 1 (TAK1) in a competitive manner with respect to ATP, displaced fluorescent ATP probe from the complex with TAK1, and docked at the ATP-binding active site on the crystal structure of TAK1. Moreover, PMOC inhibited TAK1 auto-phosphorylation, which is an axis in the activating pathways of nuclear factor-κB (NF-κB) or activating protein 1 (AP1), in the liver with ALF in vivo or in primary liver cells stimulated with TLR agonists in vitro. PMOC consequently suppressed TAK1-inducible NF-κB or AP1 activity in the inflammatory injury, an early pathogenesis leading to ALF. The results suggested that PMOC could contribute to the treatment of TLR/MyD88-associated ALF with the ATP-binding site of TAK1 as a potential therapeutic target.
Similar content being viewed by others

Introduction
Acute liver failure (ALF) is characterized by a sudden and massive death of hepatocytes with high mortality rates. Currently, liver transplantation is the only available therapeutic strategy, as pharmacological treatment of ALF patients remains elusive. Toll-like receptors (TLRs) are distributed in hepatocytes and non-parenchymal cells of the liver, and sense not only pathogen-associated molecular patterns from microbes but also danger-associated molecular pattern molecules from dying host cells, which suggests TLRs have important roles in the hepatic homeostasis to disease.1, 2 In particular, myeloid differentiation protein 2 (MD-2) in complex with TLR4 recognizes the lipid A moiety of bacterial lipopolysaccharide (LPS), while TLR9 is an endosomal receptor for microbial double-stranded (ds) DNA or oligodeoxynucleotide containing unmethylated CpG motif (CpG ODN).3, 4 CpG ODN mimics the bacterial or viral unmethylated CpG DNA that stimulates mammalian TLR9s.4 TLRs transmit immune responses cross the plasma membrane via differentiation factor 88 (MyD88) or Toll/IL-1 receptor-containing adaptor inducing IFN-β (TRIF) that are intracellular adaptor molecules.5 MyD88 is involved in several TLRs (1/2, 2/6, 4, 5, 7, 8, 9)-mediated immune processes, while TRIF is specific to TLR3 and 4.5, 6, 7
TLR/MyD88-associated immune process sequentially stimulates the phosphorylation of IL-1 receptor-associated kinase 4 (IRAK-4), the degradation of IRAK-1, and the auto-phosphorylation (activation) of TGF-β-activated kinase 1 (TAK1).5, 6 In turn, TAK1 phosphorylates the inhibitory κB (IκB) kinase (IKK) or stimulates the mitogen-activated protein kinase JNK or p38 in the activating pathways of nuclear factor-κB (NF-κB) and activating protein 1 (AP1), which upregulates expression of inflammatory genes encoding TNF-α, IL-1α, IL-6, HMGB-1 and chemokines.8, 9 On the other hand, TLR/TRIF-associated immune process triggers expression of IFN-β or IP-10 gene via the interferon regulatory factor 3 (IRF3), in which TNF receptor (TNFR)-associated factor family member-associated NF-κB activator-binding kinase 1 (TBK-1) activates IRF3 through specific phosphorylation.5, 10
LPS- or CpG ODN-induced ALF in D-galactosamine (GalN)-sensitized mice has been used to investigate the associated inflammatory and apoptotic injury mechanisms, in which GalN is exclusively metabolized to UDP-GalN in the liver, leading to UTP depletion.11, 12, 13 Pathogenesis of the immune-mediated ALF is initiated by TLR/MyD88-mediated signal cascade that elevates the production of inflammatory cytokines in non-parenchymal immune cells of the liver including Kupffer cells.14, 15 Among the hepatic cytokines, TNF-α plays important roles in the apoptosis of hepatocytes and also in the recruitment of leukocytes to liver sinusoids.16, 17 Neutrophils transmigrate into the liver to remove apoptotic cells but their exaggerated responses attack healthy hepatocytes, resulting in severe liver injury to shock-mediated death.16, 18
Chalcone derivatives are enriched in numerous medicinal plants. They exhibit hepatoprotective activities against LPS-induced ALF in GalN-sensitized mice, CCl4-induced hepatic inflammation in rats, ischemia/reperfusion-induced hepatotoxicity in mice, or hepatitis C virus-infected liver steatosis and fibrosis in dogs.19, 20, 21, 22 However, their mechanisms remain to be clarified. In the current study, we focused on molecular basis of piperidylmethyloxychalcone (PMOC, Figure 1a) in the treatment of TLR/MyD88-associated ALF in mice, and proposed the ATP-binding active site of TAK1 as a potential therapeutic target.

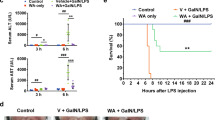
Effect of piperidylmethyloxychalcone (PMOC) on immune-mediated acute liver failure (ALF) in mice. (a) Chemical structure of PMOC. (b) C57BL/6J mice were intraperitoneally injected with lipopolysaccharide (LPS) (10 μg kg−1), CpG ODN (20 μg kg−1), GalN (500 mg kg−1), LPS (10 μg kg−1)/GalN (500 mg kg−1) or CpG ODN (20 μg kg−1)/GalN (500 mg kg−1). Survival rates were examined until 42 h after each challenge. (c, d) C57BL/6J mice were intraperitoneally intoxicated with LPS (10 μg kg−1)/GalN (500 mg kg−1) or CpG ODN (20 μg kg−1)/GalN (500 mg kg−1) for induction of ALF, and then treated with vehicle, PMOC or silymarin intravenously. Survival rates were examined until 42 h after each challenge. Data are mean±s.d. (n=12). *P<0.05 vs LPS/GalN- or CpG ODN/GalN alone-injected mice. (e) Hepatic lobules were embedded in paraffin, sectioned with 3 μm thickness, and stained with H&E for histological examination. The scale bars are 50 μm. Data are mean±s.d. (n=5). #P<0.05 vs vehicle alone-injected mice. *P<0.05 vs LPS/GalN alone-injected mice.
Materials and methods
Materials
PMOC (>97% purity) was prepared as described in Supplementary Figure S1. TLR agonists including CpG ODN were purchased from Invitrogen (Carlsbad, CA, USA), rhTAK1-TAB1 or other protein kinases from SignalChem (Richimond, Canada), enzyme-linked immunosorbent assay (ELISA) kits and cytokines (TNF-α, IL-1α, HMGB-1) from R&D Systems (Minneapolis, MN, USA), and 2′,3′-O-(2,4,6-trinitrophenyl)-adenosine triphosphate (TNP-ATP) from Life Technology (Bangalore, India). Primary and secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA) or BioLegend (San Diego, CA, USA). Escherichia coli LPS and other materials were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise specified.
Mouse ALF model
C57BL/6J male mice, aged 5–6 weeks, were purchased from KRIBB (Cheongju, Korea), and kept at the animal facility with temperature (22±2 °C), humidity (55±5%) and a 12 h/12 h light–dark cycle. Animal study was conducted in accordance with the protocol approved by the Animal Experimentation Ethics Committee in Chungbuk National University (permit number CBNUA-809-15-01). C57BL/6J mice were intraperitoneally (i.p.) challenged with E. coli LPS (10 μg kg−1) plus GalN (500 mg kg−1) (LPS/GalN) or CpG ODN (20 μg kg−1) plus GalN (500 mg kg−1) (CpG ODN/GalN) for induction of immune-mediated ALF, and then treated with vehicle or PMOC intravenously (i.v.). Survival rates of ALF-induced mice were examined as primary outcome. Blood samples were collected to analyze aminotransferase (AST, ALT) or bilirubin levels. Hepatic lobules were fixed with 10% p-formaldehyde. After dehydration, the specimens were embedded in paraffin, sectioned with 3 μm thickness and incubated with either hematoxylin and eosin (H&E) or terminal deoxynucleotidyl transferase-dUTP nick and labeling (TUNEL) assay kit (Promega, Madison, WI, USA) for histological examinations. Hepatic injury in H&E staining was scored as normal=1, mild=2, moderate=3 and severe=4. Liver homogenates were loaded onto Caspase-Glo assay kits (Promega) in the determination of caspase-3/7 or -8 activity.
Isolation of primary liver cells and macrophages
Primary liver cells were isolated from C57BL/6J mice. Mouse livers were perfused in situ via the portal vein with Ca2+-free HBSS for 10 min (flow rate 3–8 ml min−1 at 37 °C), and then digested with Liberase (30 μg ml−1 in HBSS supplemented with 5 mM CaCl2) for 10 min (flow rate 5 ml min−1 at 37 °C). Thereafter, the livers were mechanically dispersed in ice-cold HBSS supplemented with 1% BSA, filtered through an 100-μm nylon mesh, and centrifuged for 5 min at 50 g and 4 °C. The cells in the supernatant were used as whole liver cells or subjected to Percoll gradient for enrichment of Kupffer cells or hepatocytes. Whole liver cells were loaded on a 25–50% Percoll gradient and centrifuged for 30 min at 1350 g and 4 °C. Cells between the cushions were seeded into Petri dish containing RPMI medium supplemented with 10% FBS and antibiotic cocktail, and incubated for 30 min at 37 °C. After washing gently with RPMI medium, adherent cells were used as Kupffer cells. In another, whole liver cells were loaded onto 40% Percoll gradient and centrifuged for 10 min at 200 g and 4 °C. Cells in the interface were used as primary hepatocytes. The primary liver cells were maintained in type IV collagen (0.1 mg ml−1)-coated culture plates containing RPMI medium supplemented with 10% FBS and antibiotic cocktail under an atmosphere of 37 °C and 5% CO2.
RAW 264.7 monocytic cells were purchased from American Tissue Culture Collection (Manassas, VA, USA), and peritoneal macrophages were isolated from C57BL/6J mice. In brief, abdominal areas were disinfected with 70% ethyl alcohol and peritoneal cavities were flushed with ice-cold PBS to harvest macrophages. RAW 264.7 cells or peritoneal macrophages were seeded into DMEM supplemented with 10% FBS and antibiotic cocktail, and incubated under an atmosphere of 37 °C and 5% CO2.
Flow cytometric analysis
After Fc receptor blockade, cells were incubated with PE-conjugated anti-CD11b (clone: M1/70) or APC-conjugated anti-Ly6G (clone: 1A8) antibody, and subjected to flow cytometric analysis.
Reverse transcription-polymerase chain reaction
Reverse transcription-polymerase chain reaction analysis was carried out to determine mRNA levels of CCL2, CCL25, CCR2, CCR9, CXCL1, CXCL2, CXCR2, HMGB-1, IFN-β, IL-1α, IL-6 or TNF-α gene with β-actin as an internal control. Nucleotide sequences of the reverse transcription-polymerase chain reaction primers are described in Supplementary Table S1. Briefly, total RNAs (1 μg) were reversely transcribed with oligo-dT primer for 1 h at 42 °C. Single-stranded (ss) cDNAs were subjected to PCR for 27–32 cycles, in which one cycle was consisted with heat denaturation for 30 s at 94 °C, primer annealing for 30 s at 52–57 °C and DNA synthesis for 1 min at 72 °C. The reaction mixtures were resolved on agarose gels by electrophoresis and incubated with 1% ethidium bromide to visualize mRNA levels of each gene.
Western blot analysis
Cell extracts were boiled for 10 min in the presence of 0.5% SDS and 1% 2-mercaptoethanol, and resolved on acrylamide gels by electrophoresis. Proteins on the gels were transferred to a polyvinylidene difluoride membrane. Blots were blocked with 5% non-fat milk or 5% BSA in TTBS, and reacted with primary antibody for overnight at 4 °C followed by horseradish peroxidase-labeled secondary antibody for 2–4 h. After washing with TTBS, blots were soaked in an enhanced chemiluminescence reagent (GE Healthcare, Chalfont, St Giles, UK) to visualize immune complex.
In vitro kinase assay
Catalytically active rhTAK1-TAB1 or other protein kinases were reacted with major basic protein (MBP, 0.33 mg ml−1) as an exogenous substrate and [γ-32P]ATP (5 μCi) as the probe for 30 min at 30 °C. The reaction mixtures were spotted onto a P81 phosphocellulose filter, and washed extensively with 0.8% H3PO4 followed by 98% acetone. Radioactivity on the filter was measured as count per min (cpm). rhTAK1-TAB1-catalyzed kinetic parameters, Michaelis–Menten Km constant and maximal velocity (Vmax), were determined by the Lineweaver–Burk plots.
Fluorescence analysis
Fluorescent ATP probe (TNP-ATP, 2 μM) was incubated with rhTAK1-TAB1 (100 ng) for 2 h in cell-free reactions. Fluorescence values were measured as relative fluorescence units (RFUs) with excitation at 400 nm and emission at 500–610 nm.
Molecular docking
The crystallographic structure of rhTAK1-TAB1 was obtained from Protein Data Bank (code no. 4L3P), in which amino-acid sequences between human and mouse TAK1s are identical in a range of 91%. Chemical structure of PMOC was drawn in accordance with the Tripos force field and Gasteiger–Huckel charge in the Sybyl package. PMOC docking to the crystal structure of TAK1-TAB1 was simulated using the Surflex–Dock program in Sybyl package.
Confocal microscopy
Cells were fixed with 4% p-formaldehyde and permeabilized in 0.5% Triton X-100. After washing with PBS, cells were blocked with 1% BSA for 1 h, and reacted with anti-NF-κB p65 antibody for 2 h followed by Alexa Fluor 568-labeled secondary antibody for another 1 h in the dark. The cells were then incubated with 4,6-diamidino-2-phenylindole (DAPI, 3 μM) for 3 min in the dark, and examined using confocal microscopy.
Luciferase reporter assay
Luciferase reporter constructs of TNF-α (−1260/+60)-Luc or IL-1α (−1856/+1)-Luc were used in the determination of promoter activity of TNF-α or IL-1α gene,23, 24 and those of NF-κB-Luc, AP1-Luc or IRF3-Luc in the analysis of NF-κB, AP-1 or IRF3 transcriptional activity (Promega). Cells were incubated with Lipofectamine 2000 (Invitrogen) in the presence of each reporter construct and Renilla control vector. Firefly luciferase activity, a reporter, was measured as relative luminescence units and normalized to the Renilla activity, a transfection efficiency.
Secretory alkaline phosphatase reporter assay
RAW 264.7 cells that stably harbor NF-κB-secretory alkaline phosphatase (SEAP) reporter construct were used in the determination of NF-κB transcriptional activity.25 Supernatants in the cell culture were heated for 10 min at 65 °C and incubated with 4-methylumbelliferyl phosphate (400 μM) for 1 h in the dark. SEAP activity, a reporter, was measured as RFU with excitation at 360 nm and emission at 450 nm.
Statistical analysis
Data were statistically analyzed by the Dunnett’s test in one-way ANOVA program, are represented as mean±s.d. P<0.05 was considered as significantly different.
Results
PMOC rescues ALF-induced mice
LPS/GalN- or CpG ODN/GalN-injected mice were time-dependently died as a result of ALF, while LPS-, CpG ODN- or GalN alone-injected mice did not (Figure 1b). C57BL/6J mice were intoxicated with LPS/GalN or CpG ODN/GalN (i.p.) for induction of ALF, and then treated with vehicle, PMOC or silymarin (i.v.). Silymarin, a hepatoprotective drug that contains flavolignans, was used as a positive control agent.26 Treatment with PMOC or silymarin decreased mortality rates of LPS/GalN-challenged mice with ALF, such that about 70% of the mice survived in the PMOC (100 mg kg−1)- or silymarin (50 mg kg−1)-treated group, while most of them died in the vehicle alone-treated group (Figure 1c). Moreover, treatment with PMOC dose-dependently decreased ALF-associated death of CpG ODN/GalN-challenged mice (Figure 1d). LPS/GalN-challenged mice drastically increased aminotransferases (AST, ALT) or bilirubin levels in the blood (Supplementary Table S2), which are known as biochemical markers of severe liver injury. Treatment with PMOC decreased AST, ALT or bilirubin levels in the blood of ALF-induced mice (Supplementary Table S2). Concurrently, PMOC ameliorated LPS/GalN-induced liver injury, especially congestion and parenchymal degeneration in the hepatic lobules, as did silymarin as a positive control agent (Figure 1e).
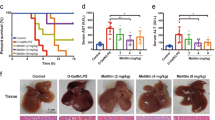
LPS/GalN- or CpG ODN/GalN-induced ALF pathogenesis stimulates hepatic inflammation and apoptotic death of hepatocytes.11, 15, 16 C57BL/6J mice were intoxicated with LPS/GalN or CpG ODN/GalN (i.p.) for induction of ALF, and then treated with vehicle or PMOC (i.v.). Treatment with PMOC decreased leukocyte infiltration into the livers of LPS/GalN-challenged mice with ALF, as evident in flow cytometric analysis with CD11b identifying immune cells such as monocytes, macrophages and neutrophils or with Ly6G as a marker of neutrophils (Figure 2a). Concurrently, PMOC suppressed mRNA levels of chemokines and their receptors in the liver with ALF (Figure 2b). Activated Kupffer or immune cells in the liver secrete CCL2 and CCL25, chemokines that recruit monocytes/macrophages via the respective receptors CCR2 and CCR9 for hepatic inflammation, or CXCL1 and CXCL2 that activate neutrophils via the common receptor CXCR2.27 TUNEL and caspase assays were then carried out to understand the apoptotic death of hepatocytes in immune-mediated ALF. Treatment with PMOC also decreased TUNEL-positive cells in the livers of CpG ODN-challenged mice with ALF (Figure 2c) as well as caspase-3/7 or -8 activity in the tissues (Figure 2d and e). The results in vivo suggested that PMOC could sequentially contribute to the amelioration of hepatic inflammation, apoptosis of hepatocytes, severe liver injury and shock-mediated death in ALF-induced mice.

Effects of piperidylmethyloxychalcone (PMOC) on leukocyte infiltration and apoptotic injury in the livers of acute liver failure (ALF)-induced mice. C57BL/6J mice were intraperitoneally intoxicated with lipopolysaccharide (LPS) (10 μg kg−1)/GalN (500 mg kg−1) or CpG ODN (20 μg kg−1)/GalN (500 mg kg−1) for induction of ALF, and then treated with vehicle or PMOC intravenously. (a) Total cells were isolated from the liver. After Fc receptor blockade, cells were stained with PE-conjugated anti-CD11b antibody or APC-conjugated anti-Ly6G antibody, and subjected to flow cytometric analysis. Data are mean±s.d. (n=5). (b) Total RNAs were isolated from liver tissues, and subjected to RT-PCR to determine mRNA levels of CCL2, CCR2, CCL25, CCR9, CXCL1, CXCL2 or CXCR2 gene with β-actin as an internal control. Data are mean±s.d. (n=3). (c) Hepatic lobules were embedded in paraffin, sectioned with 3 μm thickness, and stained with TUNEL assay kit for histological examination. The scale bars are 50 μm. Data are mean±s.d. (n=5). (d, e) Liver homogenates were loaded onto Caspase-Glo assay kits to determine caspase-3/7 or -8 activity. Data are mean±s.d. (n=5). #P<0.05 vs vehicle alone-injected mice. *P<0.05 vs LPS/GalN- or CpG ODN/GalN alone-injected mice.
PMOC suppresses NF-κB/AP1-regulated gene expression in the liver with ALF
To determine whether PMOC affected TLR-associated immune process, we examined the active indexes via specific phosphorylation of NF-κB p65 at the Ser-536 residue, c-Fos at the Ser-32 residue, c-Jun at the Ser-63 residue or IRF3 at the Ser-396 residue.28, 29 LPS/GalN-challenged mice with ALF stimulate the phosphorylation of NF-κB p65, c-Jun or IRF3 in the liver.30 CpG ODN/GalN-injected mice with ALF also markedly increased phosphor (p)-NF-κB p65 or p-c-Fos levels in the liver, while CpG ODN-, GalN- or PMOC alone-injected mice did not show any activation of the transcription factors (Supplementary Figure S2a). C57BL/6J mice were intoxicated with LPS/GalN or CpG ODN/GalN (i.p.) for induction of ALF, and then treated with vehicle or PMOC (i.v.). Treatment with PMOC decreased LPS/GalN- or CpG ODN/GalN-induced p-NF-κB p65, p-c-Fos or p-c-Jun levels in the liver (Figure 3a), which are TLR/MyD88-dependent responses. However, treatment with PMOC did not decrease LPS/GalN-induced p-IRF3 levels in the liver (Supplementary Figure S2b), suggesting no effect on TLR4/TRIF-associated immune process. Concurrently, PMOC inhibited LPS/GalN- or CpG ODN/GalN-stimulated auto-phosphorylation (activation) of TAK1 at the Thr-184 and Thr-187 residues in the liver but not that of IRAK-4 at the Thr-345 and Ser-346 residues (Figure 3b). TAK1 is located downstream from IRAK-4 in the TLR/MyD88-dependent activating pathways of NF-κB and AP1.6, 8 Furthermore, treatment with PMOC-suppressed protein and mRNA levels of NF-κB/AP1-target genes encoding TNF-α and IL-1α in the liver (Figure 3c–e), but not mRNA levels of IRF3-target IFN-β gene (Figure 3e). The results in vivo suggested that PMOC could inhibit the autophosphorylation (activation) of TAK1 in the liver with immune-mediated ALF, thus suppressing the transcriptional activity of NF-κB or AP1.

Effects of piperidylmethyloxychalcone (PMOC) on the phosphorylation (activation) of NF-κB or AP1 and the expression of TNF-α or IL-1α in the livers of acute liver failure (ALF)-induced mice. (a) C57BL/6J mice were intraperitoneally intoxicated with lipopolysaccharide (LPS) (10 μg kg−1)/GalN (500 mg kg−1) or CpG ODN (20 μg kg−1)/GalN (500 mg kg−1) for induction of ALF, and then treated with vehicle or PMOC intravenously. (a, b) Cell extracts were prepared from liver tissues, and subjected to western blot analysis (WB) with paired antibodies against p-NF-κB p65 and total NF-κB p65, p-c-Fos and total c-Fos, p-c-Jun and total c-Jun (a), p-IRAK-4 and total IRAK-4 or p-TAK1 and total TAK1 (b). Data are mean±s.d. (n=3). (c, d) Liver homogenates were loaded onto ELISA kits to determine protein levels of TNF-α or IL-1α. Data are mean±s.d. (n=5). (e) Total RNAs were prepared from liver tissues, and subjected to RT-PCR to determine mRNA levels of TNF-α, IL-1α or IFN-β gene with β-actin as an internal control. Data are mean±s.d. (n=3). #P<0.05 vs vehicle alone-injected mice. *P<0.05 vs LPS/GalN- or CpG ODN/GalN alone-injected mice.
PMOC inhibits TAK1 auto-phosphorylation in primary liver cells and macrophages
PMOC did not affect LPS binding to rhMD-2 in cell-free reactions, whereas lipid IVa (MD-2 antagonist) as a positive control agent resulted in inhibition of the binding (Supplementary Figure S3a), thereby excluding the possibility of direct effect of PMOC on the receptor or LPS scavenging. To elucidate a mechanism of PMOC, we examined TAK1 auto-phosphorylation in primary liver cells, because it decreased p-TAK1 levels in the livers of ALF-induced mice shown in Figure 3b. PMOC inhibited TAK1 auto-phosphorylation in LPS- or CpG ODN-activated whole liver cells (Figure 4a). PMOC also decreased Pam3CSK4 (TLR1/2 agonist mimicking the triacylated lipoprotein from Gram-positive bacteria)-, flagellin (TLR5 agonist from bacterial flagellar filament)- or ssRNA (TLR7 agonist mimicking the viral RNA)-induced p-TAK1 levels in whole liver cells (Figure 4b), as well as TNF-α-, IL-1α- or HMGB-1-induced p-TAK1 levels in the cells (Figure 4c). Moreover, PMOC inhibited TAK1 auto-phosphorylation in LPS- or CpG ODN-activated Kupffer cells and TNF-α- or IL-1α-activated hepatocytes, in which LLZ 1640-2 was used as a positive control agent (Figure 4d and e). LLZ 1640-2 is an irreversible ATP-competitive inhibitor of TAK1-catalyzed kinase activity.31 Similarly, PMOC inhibited TAK1 auto-phosphorylation in LPS-, CpG ODN- or TNF-α-activated mouse peritoneal macrophages and RAW 264.7 cells (Supplementary Figure S3b and c). PMOC at concentrations up to 30 μM did not disturb the viability of whole liver cells (Supplementary Figure S4a), thereby excluding the possibility of nonspecific cytotoxicity.

Effect of piperidylmethyloxychalcone (PMOC) on TAK1 auto-phosphorylation in primary liver cells. Primary liver cells were isolated from C57BL/6J mice. Whole liver cells were pretreated with PMOC for 2 h and stimulated with lipopolysaccharide (LPS) or CpG ODN (a), other TLR agonists (b) or cytokines (c) for 10–20 min in the presence of PMOC. (d) Kupffer cells were pretreated with PMOC for 2 h and stimulated with LPS or CpG ODN for 10–20 min in the presence of PMOC. (e) Hepatocytes were pretreated with PMOC for 2 h and stimulated with TNF-α or IL-1α for 10–20 min in the presence of PMOC. Cell extracts were subjected to western blot analysis (WB) with anti-p-TAK1 or anti-TAK1 antibody. Data are mean±s.d. (n=3). #P<0.05 vs media alone-added group. *P<0.05 vs each TLR agonist- or each cytokine alone-stimulated group.
On the other hand, PMOC affected neither the IRAK-4 auto-phosphorylation in LPS- or CpG ODN-activated whole liver cells and RAW 264.7 cells, in which N-(2-morpholinylethyl)-2-(3-nitrobenzoylamindo)-benzimidazole (IS409, IRAK-1/4 inhibitor) was used as a positive control agent (Supplementary Figure S4b and c), nor the IRAK-1 degradation in LPS-activated cells (Supplementary Figure S4d). The results in vitro suggested that PMOC could inhibit TAK1 auto-phosphorylation in primary liver cells and macrophages stimulated with various TLR agonists or endogenous cytokines, while it had no effects on TLR/MyD88-recruited early signal transducers, including IRAK-1 and -4, that are located upstream from TAK1.
PMOC is an ATP-competitive inhibitor of TAK1-catalyzed kinase activity
To determine whether PMOC directly inhibited the kinase activity of TAK1, catalytically active rhTAK1-TAB1 was treated with PMOC in cell-free reactions and reacted with MBP as an exogenous substrate in the presence of [γ-32P]ATP probe. PMOC dose-dependently inhibited the rhTAK1-TAB1-catalyzed kinase activity, as did LLZ 1640-2 as a positive control agent (Figure 5a). In a kinetic study, rhTAK1-TAB1 exhibited a Km of 0.09 μM and a Vmax of 1570 Δcpm min−1 with varying concentrations of ATP (Figure 5b). Treatment with PMOC increased the Km value but did not alter the Vmax value of rhTAK1-TAB1-catalyzed kinase activity (Figure 5b), suggesting a competitive inhibitor with respect to ATP. However, PMOC did not inhibit the catalytic activities of cell-free rhIRAK-4, rhIKKβ, rhJNK and rhTBK-1 that are also involved in TLRs-associated immune responses, in which IS409 (IRAK-1/4 inhibitor), sulfasalazine (IKKβ inhibitor), SP 600125 (JNK inhibitor) and amlexanox (TBK-1 inhibitor) were used as positive control agents (Figure 5c–f).

Effect of piperidylmethyloxychalcone (PMOC) on TAK1-catalyzed kinase activity. Catalytically active rhTAK1-TAB1 (a, b), rhIRAK-4 (c), rhIKKβ (d), rhJNK (e) or rhTBK-1 (f) was treated with PMOC for 10 min in cell-free reactions. In vitro kinase activity was then monitored by incorporation of [32P] from the [γ-32P]ATP probe onto MBP as an exogenous substrate. Data are mean±s.d. (n=5). *P<0.05 vs rhTAK1-TAB1- or other protein kinase alone-containing group. (b) Kinetic data of rhTAK1-TAB1-catalyzed kinase activity are represented as 1/V, an inverse of the initial increase of cpm values per min (Δcpm min−1), with varying concentrations of ATP.
To clarify whether PMOC interacted with the ATP-binding site of TAK1, fluorescent ATP probe (TNP-ATP) was pre-incubated with rhTAK1-TAB1 in cell-free reactions to achieve stable fluorescence intensity, and treated with PMOC. The fluorescence intensity of TNP-ATP was markedly increased following its binding to rhTAK1-TAB1 (Figure 6a). Post treatment with PMOC decreased rhTAK1-TAB1-enhanced fluorescence intensity of TNP-ATP (Figure 6a), indicating displacement of TNP-ATP from the complex with rhTAK1-TAB1. However, non-fluorescent PMOC did not alter basal fluorescence values of TNP-ATP in the absence of rhTAK1-TAB1 (Figure 6b). LLZ 1640-2 as a positive control agent also displaced TNP-ATP from the complex with rhTAK1-TAB1, as did PMOC (Figure 6b).

Effect of piperidylmethyloxychalcone (PMOC) on displacement of fluorescent ATP probe from the complex with rhTAK1-TAB1. (a) Fluorescent ATP probe (TNP-ATP, 2 μM) was pre-incubated with rhTAK1-TAB1 (100 ng) for 2 h in cell-free reactions to achieve stable fluorescence intensity under excitation at 400 nm, and treated with PMOC for another 2 h. Emission spectra at 500–610 nm are represented as relative fluorescence units (RFUs). (b) TNP-ATP was pre-incubated in the absence or presence of rhTAK1-TAB1 for 2 h and treated with PMOC for another 2 h. Fluorescence intensity was measured as RFU under excitation at 400 nm and emission at 550 nm. Data are mean±s.d. (n=5). #P<0.05 vs TNP-ATP alone-containing group. *P<0.05 vs TNP-ATP plus rhTAK1-TAB1 alone-containing group. (c) Docking arrangement of PMOC to the crystal structure of TAK1 was carried out with the Surflex-Dock program. PMOC is represented as a green color, and TAK1 as a grey color. Hydrogen bonding between PMOC and TAK1 is indicated as a black dotted line. (d) Superimposed docking of PMOC onto the endogenous ligand, ATP bound to TAK1. PMOC is represented as a green color, ATP as a yellow color, and TAK1 as a gray color.
Based on the evidences that PMOC competitively inhibited the kinase activity of rhTAK1-TAB1 with varying concentrations of ATP and displaced fluorescent ATP probe from the complex with rhTAK1-TAB1, we conducted molecular docking with the crystal structure of human TAK1.32 PMOC was docked at the ATP-binding active site of TAK1 consisting with the Glu-105, Ala-107 and Asn-114 residues in the most favorable simulation (Figure 6c). Hydrogen bonding was achieved between the 2′-hydroxyl group of PMOC and the peptidyl carbonyl group of Glu-105 or the peptidyl amino group of Ala-107, and between the 4-hydroxyl group of PMOC and the side chain of Asn-114 (Figure 6c). Moreover, the docking simulation of PMOC overlapped with that of the endogenous ligand, ATP bound to TAK1 (Figure 6d).
PMOC inhibits TAK1-inducible transcriptional activity of NF-κB or AP1
The inhibition of cellular TAK1 could affect TLR/MyD88- or TNFR-dependent activating pathways of NF-κB and AP1, because TAK1 is located upstream from the pathways.5, 8, 9 Treatment with PMOC sequentially inhibited IκBα phosphorylation in LPS- or CpG ODN-activated whole liver cells and LPS-activated RAW 264.7 cells (Supplementary Figure S5a and b), nuclear import of NF-κB p65 in LPS-activated RAW 264.7 cells (Supplementary Figure S5c), and NF-κB transcriptional activity in LPS-, CpG ODN- or TNF-α-activated cells containing NF-κB-Luc reporter construct (Supplementary Figure S5d). To determine whether TAK1 was a primary target of PMOC in the inhibition of NF-κB activating pathway, RAW 264.7 cells that stably harbor NF-κB-SEAP reporter construct were transfected with expression vector of TAK1-TAB1 or IKKβ. The ectopic expression of TAK1-TAB1 or IKKβ, which bypasses the TLRs or cytokine receptors, increased NF-κB transcriptional activity that was reported by SEAP activity (Supplementary Figure S5e and f). PMOC inhibited TAK1-TAB1 vector-elicited NF-κB transcriptional activity (Supplementary Figure S5e), but not SEAP activity under the control of IKKβ vector (Supplementary Figure S5f), thereby excluding the possibility of direct effect of POMC on the NF-κB activating pathway that is located downstream from TAK1.
PMOC also inhibited AP1-activating pathway in primary liver cells, including the LPS- or CpG ODN-induced phosphorylation of JNK1/2 at the Thr-183 residue or p38 at the Thr-180 residue (Supplementary Figure S6a) and the LPS-induced phosphorylation of c-Fos or c-Jun, in which LLZ 1640-2 and SP 600125 were used as positive control agents (Supplementary Figure S6b). Moreover, PMOC decreased p-c-Fos or p-c-Jun levels in the nucleus of LPS-, CpG ODN- or TNF-α-activated RAW 264.7 cells (Supplementary Figure S6c), and inhibited AP1 transcriptional activity in LPS- or CpG ODN-activated cells containing AP1-Luc reporter construct (Supplementary Figure S6d).
On the other hand, PMOC affected neither the IRF3 phosphorylation in LPS-activated whole liver cells and poly I:C (TLR3 agonist)- or LPS-activated RAW 264.7 cells (Supplementary Figure S7a and b), nor the IRF3 transcriptional activity in LPS-activated cells containing IRF3-Luc reporter construct (Supplementary Figure S7c). The results in vitro suggested that PMOC interrupted TAK1-inducible transcriptional activity of NF-κB or AP1 in primary liver cells and macrophages, whereas it had no effects on TLR/TRIF-dependent IRF3 activity.
PMOC suppressed protein and mRNA levels of NF-κB/AP1-target genes encoding TNF-α, IL-1α, IL-6 or HMGB-1 in LPS-activated whole liver cells (Supplementary Table S3; Supplementary Figure S8a), which was consistent with the in vivo effects in ALF-induced mice shown in Figure 3c–e. RAW 264.7 cells were then transfected with either TNF-α-Luc reporter construct containing the TNF-α promoter region (−1260/+60) or IL-1α-Luc reporter construct containing the IL-1α promoter region (−1856/+1). PMOC inhibited LPS- or CpG ODN-stimulated promoter activity of TNF-α gene that was reported by firefly luciferase assay (Supplementary Figure S8b), as wells as LPS- or TNF-α-stimulated promoter activity of IL-1α gene (Supplementary Figure S8c), suggesting that PMOC could downregulate the expression of NF-κB/AP1-target genes at the transcription level. However, PMOC did not affect protein levels of IRF3-target genes encoding IFN-β and IP-10 in LPS-activated whole liver cells (Supplementary Table S3).
Discussion
In the current study, PMOC ameliorated LPS- or CpG ODN-induced ALF in GalN-sensitized C57BL/6J mice via suppressing TLR/MyD88-dependent immune process. As a molecular basis, PMOC directly inhibited TAK1-catalyzed kinase activity in a competitive mechanism with respect to ATP, and displaced fluorescent ATP probe from the complex with TAK1-TAB1. Consistent with these findings, PMOC was docked at the ATP-binding active site on the crystal structure of TAK1 in the most favorable simulation.
The importance of TLR4, TLR9, or MyD88 in hepatic diseases has been demonstrated in mice with gene knockout (KO). TLR4-KO mice are completely resistant to LPS/GalN-induced ALF, and relatively hypo-responsive to acetaminophen-induced liver necrosis and multi-organ dysfunction compared with the wild-type strains.33, 34 TLR4-KO mice are also protected from ischemia/reperfusion-induced hepatotoxicity and alcohol-induced hepatic inflammation.35, 36 TLR9-KO mice are insensitive to the CpG ODN/GalN-induced ALF with suppressed expression of NF-κB/AP1-target genes encoding TNF-α, IL-1β or IL-6, and to the acetaminophen- or concanavalin A-induced ALF.12, 37, 38 MyD88-KO mice elude ALF induced by LPS following the sensitization with gut-resident Propionibacterium acnes, in which MyD88 in non-parenchymal immune cells rather than hepatocytes is critically involved in the associated inflammatory injury.36, 39 Moreover, MyD88-KO mice are attenuated from diethylnitrosamine-induced hepatocarcinogenesis, bile duct ligation-induced hepatic fibrosis, and malaria Plasmodium berghei-infected liver damage.40, 41, 42
LLZ 1640-2 is well known as a selective inhibitor of TAK1, and was used as a positive control agent in this study. LLZ 1640-2 has an unsaturated cis-enone moiety that attacks the Cys-174 residue in ATP-binding active site of TAK1 to make a covalent adduct.43 LLZ 1640-2 is rapidly inactivated in human or mouse microsomes and plasma due to cis-to-trans isomerization of the enone moiety via Michael addition/elimination with glutathione (GSH) and glutathione transferase (GST) in biological fluids.44 Moreover, the trans-isomer of LLZ 1640-2 is 50-fold less potent than the parent cis-isomer in the inhibition of LPS-induced TNF-α promoter activity in THP-1 cells.44 In the current study, PMOC inhibited TAK1-catalyzed kinase activity via a reversible competition with respect to ATP, suggesting that its unsaturated enone moiety might be dispensable in the molecular mechanism of action. However, in vivo metabolism and pharmacokinetics of PMOC remain to be determined.
Taken together, we propose the ATP-binding active site of TAK1 as a molecular target of PMOC in the treatment of TLR/MyD88-associated ALF in mice. Hence, PMOC inhibited the auto-phosphorylation (activation) of TAK1 in the livers of LPS/GalN- or CpG ODN/GalN-challenged mice with ALF, as well as the TAK1-catalyzed kinase activity in primary liver cells and macrophages stimulated with TLR (1/2, 4, 5, 7, 9) agonists or endogenous cytokines (TNF-α, IL-1α, HMGB-1). PMOC consequently suppressed TAK1-inducible NF-κB or AP1 activation and NF-κB/AP1-regulated gene expression in the liver, thus targeting inflammatory injury in the pathogenic pathway of ALF. Finally, this study suggested a therapeutic target of immune-mediated ALF following insult with TLR pathogens.
References
Chen Y, Sun R . Toll-like receptors in acute liver injury and regeneration. Int Immunopharmacol 2011; 11: 1433–1441.
Heymann F, Tacke F . Immunology in the liver—from homeostasis to disease. Nat Rev Gastroenterol Hepatol 2016; 13: 88–110.
Klett J, Reeves J, Oberhauser N, Perez-Regidor L, Martin-Santamaria S . Modulation of Toll-like receptor 4: insights from X-ray crystallography and molecular modeling. Curr Top Med Chem 2014; 14: 2672–2683.
Bauer S . Toll-like receptor 9 processing: the key event in Toll-like receptor 9 activation? Immunol Lett 2013; 149: 85–87.
Bryant CE, Symmons M, Gay NJ . Toll-like receptor signalling through macromolecular protein complexes. Mol Immunol 2015; 63: 162–165.
Warner N, Nunez G . MyD88: a critical adaptor protein in innate immunity signal transduction. J Immunol 2013; 190: 3–4.
Ve T, Gay NJ, Mansell A, Kobe B, Kellie S . Adaptors in toll-like receptor signaling and their potential as therapeutic targets. Curr Drug Targets 2012; 13: 1360–1374.
Ajibade AA, Wang HY, Wang RF . Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol 2013; 34: 307–316.
Napetschnig J, Wu H . Molecular basis of NF-κB signaling. Annu Rev Biophys 2013; 42: 443–468.
Ikushima H, Negishi H, Taniguchi T . The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol 2013; 78: 105–116.
Maes M, Vinken M, Jaeschke H . Experimental models of hepatotoxicity related to acute liver failure. Toxicol Appl Pharmacol 2016; 290: 86–97.
Yi AK, Yoon H, Park JE, Kim BS, Kim HJ, Martinez-Hernandez A . CpG DNA-mediated induction of acute liver injury in D-galactosamine-sensitized mice: the mitochondrial apoptotic pathway-dependent death of hepatocytes. J Biol Chem 2006; 281: 15001–15012.
Decker K, Keppler D . Galactosamine hepatitis: key role of the nucleotide deficiency period in the pathogenesis of cell injury and cell death. Rev Physiol Biochem Pharmacol 1974; 71: 77–106.
Tsutsui H, Nishiguchi S . Importance of Kupffer cells in the development of acute liver injuries in mice. Int J Mol Sci 2014; 15: 7711–7730.
Tacke F, Luedde T, Trautwein C . Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol 2009; 36: 4–12.
Bantel H, Schulze-Osthoff K . Mechanisms of cell death in acute liver failure. Front Physiol 2012; 3: 79.
Rutherford A, Chung RT . Acute liver failure: mechanisms of hepatocyte injury and regeneration. Semin Liver Dis 2008; 28: 167–174.
Dorman RB, Gujral JS, Bajt ML, Farhood A, Jaeschke H . Generation and functional significance of CXC chemokines for neutrophil-induced liver injury during endotoxemia. Am J Physiol Gastrointest Liver Physiol 2005; 288: G880–G886.
Guan LP, Nan JX, Jin XJ, Jin QH, Kwak KC, Chai KY et al. Protective effects of chalcone derivatives for acute liver injury in mice. Arch Pharm Res 2005; 28: 81–86.
Zhao Z, Park SM, Guan L, Wu Y, Lee JR, Kim SC et al. Isoliquiritigenin attenuates oxidative hepatic damage induced by carbon tetrachloride with or without buthionine sulfoximine. Chem Biol Interact 2015; 225: 13–20.
Dorn C, Massinger S, Wuzik A, Heilmann J, Hellerbrand C . Xanthohumol suppresses inflammatory response to warm ischemia-reperfusion induced liver injury. Exp Mol Pathol 2013; 94: 10–16.
Yang M, Li N, Li F, Zhu Q, Liu X, Han Q et al. Xanthohumol, a main prenylated chalcone from hops, reduces liver damage and modulates oxidative reaction and apoptosis in hepatitis C virus infected Tupaia belangeri. Int Immunopharmacol 2013; 16: 466–474.
Baer M, Dillner A, Schwartz RC, Sedon C, Nedospasov S, Johnson PF . Tumor necrosis factor α transcription in macrophages is attenuated by an autocrine factor that preferentially induces NF-κB p50. Mol Cell Biol 1998; 18: 5678–5689.
Hawn TR, Ozinsky A, Underhill DM, Buckner FS, Akira S, Aderem A . Leishmania major activates IL-1α expression in macrophages through a MyD88-dependent pathway. Microbes Infect 2002; 4: 763–771.
Moon KY, Hahn BS, Lee J, Kim YS . A cell-based assay system for monitoring NF-κB activity in human HaCat transfectant cells. Anal Biochem 2001; 292: 17–21.
Loguercio C, Festi D . Silybin and the liver: from basic research to clinical practice. World J Gastroenterol 2011; 17: 2288–2301.
Marra F, Tacke F . Roles for chemokines in liver disease. Gastroenterology 2014; 147: 577–594 e1.
Buss H, Dorrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M . Constitutive and interleukin-1-inducible phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)-α, IKKβ, IKKɛ, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem 2004; 279: 55633–55643.
Chen W, Srinath H, Lam SS, Schiffer CA, Royer WE Jr, Lin K . Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. J Mol Biol 2008; 379: 251–260.
Park SH, Baek SI, Yun J, Lee S, Yoon da Y, Jung JK et al. IRAK4 as a molecular target in the amelioration of innate immunity-related endotoxic shock and acute liver injury by chlorogenic acid. J Immunol 2015; 194: 1122–1130.
Ninomiya-Tsuji J, Kajino T, Ono K, Ohtomo T, Matsumoto M, Shiina M et al. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J Biol Chem 2003; 278: 18485–18490.
Brown K, Vial SC, Dedi N, Long JM, Dunster NJ, Cheetham GM . Structural basis for the interaction of TAK1 kinase with its activating protein TAB1. J Mol Biol 2005; 354: 1013–1020.
Ben Ari Z, Avlas O, Pappo O, Zilbermints V, Cheporko Y, Bachmetov L et al. Reduced hepatic injury in Toll-like receptor 4-deficient mice following D-galactosamine/lipopolysaccharide-induced fulminant hepatic failure. Cell Physiol Biochem 2012; 29: 41–50.
Shah N, Montes de Oca M, Jover-Cobos M, Tanamoto K, Muroi M, Sugiyama K et al. Role of Toll-like receptor 4 in mediating multiorgan dysfunction in mice with acetaminophen induced acute liver failure. Liver Transpl 2013; 19: 751–761.
Uchida Y, Ke B, Freitas MC, Yagita H, Akiba H, Busuttil RW et al. T-cell immunoglobulin mucin-3 determines severity of liver ischemia/reperfusion injury in mice in a TLR4-dependent manner. Gastroenterology 2010; 139: 2195–2206.
Hritz I, Velayudham A, Dolganiuc A, Kodys K, Mandrekar P, Kurt-Jones E et al. Bone marrow-derived immune cells mediate sensitization to liver injury in a myeloid differentiation factor 88-dependent fashion. Hepatology 2008; 48: 1342–1347.
Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Lima BH et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 2012; 56: 1971–1982.
Jiang W, Sun R, Zhou R, Wei H, Tian Z . TLR-9 activation aggravates concanavalin A-induced hepatitis via promoting accumulation and activation of liver CD4+ NKT cells. J Immunol 2009; 182: 3768–3774.
Imamura M, Tsutsui H, Yasuda K, Uchiyama R, Yumikura-Futatsugi S, Mitani K et al. Contribution of TIR domain-containing adapter inducing IFN-β-mediated IL-18 release to LPS-induced liver injury in mice. J Hepatol 2009; 51: 333–341.
Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007; 317: 121–124.
Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA et al. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat Med 2007; 13: 1324–1332.
Adachi K, Tsutsui H, Kashiwamura S, Seki E, Nakano H, Takeuchi O et al. Plasmodium berghei infection in mice induces liver injury by an IL-12- and toll-like receptor/myeloid differentiation factor 88-dependent mechanism. J Immunol 2001; 167: 5928–5934.
Wu J, Powell F, Larsen NA, Lai Z, Byth KF, Read J et al. Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z-7-Oxozeaenol. ACS Chem Biol 2013; 8: 643–650.
Du H, Matsushima T, Spyvee M, Goto M, Shirota H, Gusovsky F et al. Discovery of a potent, metabolically stabilized resorcylic lactone as an anti-inflammatory lead. Bioorg Med Chem Lett 2009; 19: 6196–6199.
Acknowledgements
This study was financially supported by the 2014 research grant from Chungbuk National University, the foresting project of Osong academy-industry convergence from the Korean Ministry of Trade, Industry & Energy, and the grant 2015R1D1A1A01057043 or the MRC program from the National Research Foundation of Korea. We thank Dr PF Johnson (National Cancer Institute, Frederick, MD, USA) for kind supply of TNF-α-Luc construct, Dr A Aderem (University of Washington, Seattle, WA, USA) for IL-1α-Luc construct, and Dr YS Kim (Seoul National University, Seoul, Korea) for NF-κB-SEAP construct.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Park, S., Kwak, JA., Jung, SH. et al. Piperidylmethyloxychalcone improves immune-mediated acute liver failure via inhibiting TAK1 activity. Exp Mol Med 49, e392 (2017). https://doi.org/10.1038/emm.2017.156
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2017.156
