
Abstract
Wnt10b, an endogenous inhibitor of adipogenesis, maintains preadipocytes in an undifferentiated state by suppressing adipogenic transcription factors. We have previously demonstrated that Wnt10b transcription during adipogenesis is negatively regulated by X-box-binding protein 1 (XBP1), an important transcription factor of the unfolded protein response. In this report, we demonstrate that XBP1s can directly induce the transcription of microRNA-148a, which in turn mediates the silencing of Wnt10b mRNA during adipogenic differentiation of 3T3-L1 cells. Stability of Wnt10b mRNA was found to be significantly increased by knockdown of XBP1s. Using computational algorithms, a set of microRNAs was predicted to bind Wnt10b mRNA, of which microRNA-148a was selected as a potential target for XBP1s. Our results revealed that microRNA-148a could bind to the 3′UTR of Wnt10b mRNA. Its ectopic expression significantly suppressed both Wnt10b expression and β-catenin activity. When we altered the expression of XBP1 in 3T3-L1 cells, microRNA-148a levels changed accordingly. A potential XBP1 response element was found in the promoter region of microRNA-148a, and XBP1s directly bound to this response element as shown by point mutation analysis and chromatin immunoprecipitation assay. In addition, a microRNA-148a mimic significantly restored adipogenic potential in XBP1-deficient 3T3-L1 cells. These findings provide the first evidence that XBP1s can regulate Wnt10b by a post-transcriptional mechanism through directly inducing microRNA-148a.
Similar content being viewed by others


Introduction
Adipose tissue is crucial for lipid storage, thermogenesis and endocrine regulation of whole-body metabolism. Adipose tissue mass is flexibly regulated, and its timely expansion in response to positive energy balance is now considered a beneficial response to prevent ectopic fat accumulation and lipotoxicity.1 Adipocyte hypertrophy and hyperplasia are the two main mechanisms that contribute to adipose tissue expansion. Adipocyte hyperplasia from precursor cells (adipogenesis) is an essential process for adipose mass homeostasis that replaces lost adipocytes throughout life. Therefore, it is critical for the maintenance of metabolic homeostasis, that is, alleviation of systemic insulin resistance by replacing hypertrophic adipocytes with new and small cells. In this regard, recent findings have suggested that impaired adipogenesis is causally linked to hypertrophic obesity phenotypes and metabolic complications.1 However, the molecular mechanisms responsible for adipogenesis are complicated because numerous hormones, lipid mediators and transcription factors are involved.
The wingless-type MMTV integration site family (Wnt)/β-catenin signaling pathway is one of the most important regulators of adipogenic differentiation. Among Wnt family members, Wnt10b is the major molecular switch that strongly represses adipogenesis,2 and its involvement in the pathogenesis of obesity has also been suggested.3 However, the regulatory mechanisms of Wnt10b expression during adipogenesis remain unclear.
MicroRNAs are small noncoding RNAs that function as guide molecules in RNA silencing. Myriad evidence has shown that they are involved in a variety of physiological processes. MicroRNAs also have important roles in adipogenesis, and their possible applications as biomarkers and therapeutic targets for obesity have been suggested.4 Currently, multiple microRNAs have been shown to regulate adipogenesis. Many key molecules involved in adipogenesis have been indicated as potential target molecules for microRNAs.5 Among them, the Wnt/β-catenin signaling pathway is known to be involved in microRNA-induced regulation of adipogenesis.6, 7 However, despite a growing number of studies showing the importance of microRNAs in adipocyte development, the regulatory mechanisms for their transcription are largely unknown.
X-box-binding protein 1 (XBP1) is a member of the bZIP family of transcription factors. Its transcriptionally active isoform (XBP1s) is synthesized after IRE1α-mediated splicing of XBP1 mRNA. One of the main functions of XBP1s is to regulate the development of cells such as plasma cells,8 Paneth cells,9 pancreatic acinar cells,10 dendritic cells11 and eosinophils.12 Many reports have recently demonstrated the involvement of XBP1 in the regulation of adipogenesis.13, 14, 15 We have also reported that XBP1s has a pro-adipogenic role by transcriptional regulation of peroxisome proliferator-activated receptor γ (PPARγ)13 and Wnt10b.16 For the latter, XBP1 induces adipogenesis by directly suppressing Wnt10b transcription, and, recently, we found that Wnt10b mRNA stability could be decreased by XBP1s.
Therefore, we hypothesized that XBP1s could suppress Wnt10b through post-transcriptional mechanisms. Through this novel mechanism of adipogenesis, XBP1 directly induces microRNA-148a (miR-148a) production, resulting in decreased Wnt10b mRNA stability in 3T3-L1 cells.
Materials and methods
Cell culture and differentiation of 3T3-L1 preadipocytes
The mouse 3T3-L1 preadipocytes (American Type Culture Collection, Manassas, VA, USA) were cultured and differentiated into adipocytes as previously described.17 Briefly, cells were maintained for 2 days in high-glucose (4.5 g l−1) DMEM containing 10% NCS (Gibco, Grand Island, NY, USA). Two days after reaching confluence (day 0), adipogenic differentiation was induced for 2 days in the medium containing insulin (1 μg ml−1, Sigma, St Louis, MO, USA), IBMX (0.5 mM; Sigma) and dexamethasone (1 μM, Sigma). Then, the cells were cultured for 2 days in the DMEM with 10% fetal bovine serum (FBS; Gibco) and insulin (1 μg ml−1), and thereafter the medium was changed to the Dulbecco’s modified Eagle’s medium/10% FBS until day 8.
Nile Red staining and quantification of intracellular triacylglycerol content
After washing twice with phosphate-buffered saline, cells were fixed with 10% formalin in distilled water (DW) for 10 min, rinsed with DW and stained for 10 min with 1 μg ml−1 of Nile Red (Sigma) as previously described.18 Intracellular triacylglycerol levels were determined by using a Serum Triglyceride Determination Kit (Sigma) following the manufacturer’s protocol. Briefly, cells were washed with phosphate-buffered saline and lysed with RIPA buffer (20 mM Tris-HCl, pH 7.5, 0.1% sodium dodecylsulphate, 1% Triton X-100, 1% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1% NP-40 and protease inhibitors) and then incubated on ice for 10 min. The resulting lysates were centrifuged at 16 000 g for 10 min, and 10 μl of glycerol standard or samples was mixed with 500 μl of the reagents. Triacylglycerol concentration was calculated using the initial and final absorbances at 540 nm and normalized to total cellular protein. For statistical analysis, images of each dish were captured with an inverted microscope and analyzed using the Optimas 6.5 software (Meyer Instruments, Houston, TX, USA).
Genetic manipulation of XBP1
3T3-L1 cells in which XBP1 was knocked down (XBP1-KD) were prepared using retroviral vectors expressing short hairpin RNA targeting mouse XBP1 (SFGneo-iXBP1) as described.19 Briefly, 293T cells were transfected for 48 h with the viral vectors using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA), and virus-containing medium was collected. For infection, Polybrene (hexadimethrine bromide; 8 μg ml−1, Sigma) was added to the filter-sterilized virus-loaded medium and applied to proliferating cells for 2 days. For the preparation of 3T3-L1 cells overexpressing XBP1s (XBP1s-OE), recombinant plasmids expressing mouse XBP1s (pcDNA3.1-mXBP1s) were transfected using Lipofectamine 2000 reagent (Invitrogen). Genetically engineered cells were selected with G418 (1 mg ml−1). For transient knockdown of XBP1s, specific short interfering RNAs targeting XBP1 (siXBP1, Bioneer, Daegion, Korea) were transfected into 3T3-L1 cells using Lipofectamine RNAiMAX reagent (Invitrogen). Non-targeting control (siCON, Bioneer) was used as a negative control.
MicroRNA target site prediction
A search for predicted target mRNAs was performed using the online programs TargetScan (http://www.targetscan.org), miRDB (http://www.mirdb.org/miRDB) and microRNA.org (http://www.microrna.org).
Ectopic expression and knockdown of miR-148a
miR-148a mimic or inhibitor (Bioneer) was used to modify intracellular miR-148a levels. For transfection experiments, cells were grown in six-well tissue culture plates and transfected for 24 h using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer’s protocol.
Real-time PCR analysis
Total RNA was extracted using TRIzol reagent (Invitrogen), and complementary DNA was synthesized using a PrimeScript RT kit (TaKaRa, Shiga, Japan). The real-time PCR analysis was performed using an ABI 7300 thermal cycler (Applied Biosystems, Foster City, CA, USA) with SYBR Green Q-PCR Master Mix (TaKaRa). The ΔΔCt method was used to calculate the relative amount of mRNAs with the 36b4 as an internal control. The primer sequences are listed in Table 1.
Measurement of mature and primary miR-148a levels
Expression of the mature form of miR-148a was determined with real-time PCR analysis using the TaqMan microRNA reverse transcription kit with a probe for miR-148a as described in the manufacturer's protocol (Applied Biosystems). A probe for U6 was used as a control. The primary miR-148a level was examined using a semiquantitative reverse transcription PCR method that employs primers covering different regions around putative miR-148a precursor sequences (Figure 3c). Amplified fragments were separated using electrophoresis on a 1% agarose gel, visualized with ethidium bromide staining and detected using a Gel Doc EZ system (Bio-Rad, Hercules, CA, USA).
Immunoblot analysis
Cells were lysed in RIPA buffer, and 30 μg of protein was separated using 10% sodium dodecylsulphate-PAGE and transferred onto a nitrocellulose membrane (Schleicher & Schuell, Dassel, Germany). The membranes were incubated with mouse antisera against β-actin (1:500; Sigma), green fluorescent protein (GFP; 1:500; Santa Cruz Biotechnology, Dallas, TX, USA) and adiponectin (1:1000; Alexis, Farmingdale, NY, USA) or with rabbit antisera against XBP1 (1:200; BioLegend, San Diego, CA, USA), Wnt10b (1:500; Santa Cruz Biotechnology), C/EBPα (1:500; Santa Cruz Biotechnology) and PPARγ (1:500; Santa Cruz Biotechnology).20 Antibody binding was detected using horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (1:2,000; Sigma), and the immunoreactive bands were visualized using the ECL method (Amersham Pharmacia, Buckinghamshire, UK). For statistical analysis, immunoreactivities were quantified with densitometry using a Gel Doc EZ System (Bio-Rad).
Luciferase reporter analysis
Cells were seeded into 24-well plates at a density of 2 × 104 per well and transfected with reporter plasmids (50 ng) and effector or empty vectors (1–500 ng). After 48 h, cells were lysed and the luciferase activities were measured using a Dual Luciferase Assay Kit (Promega, Madison, WI, USA). Plasmids expressing the mouse miR-148a promoter (pGL3-mmu-miR148a promoter-Luc) and mouse Wnt10b promoter (pGL2-mWnt10b promoter-Luc) were used as reporters. To monitor the Wnt/β-catenin signaling pathway, Super TOPFlash, which encodes seven copies of LEF/TCF-binding sites linked to firefly luciferase (Addgene, Cambridge, MA, USA), was used as previously described.21 Transfection efficiency was normalized with the Renilla-expressing plasmids (pRL-CMV-luc; 5 ng).
Wnt10b 3′UTR reporter assay
HEK293T cells were first transfected with a miR-148a mimic or non-silencing control. The next day, cells were transfected with 0.5 μg of enhanced GFP (EGFP) reporter vector. After 24 h, cells were lysed in RIPA buffer for immunoblot analysis. Ten micrograms of total protein were used to measure the GFP expression level. The assay was performed in triplicate and repeated at least three times.
Site-directed mutagenesis
Binding motifs on pGL3-mmu-miR148a promoter-Luc were mutated using a QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). Mutagenesis was carried out by PCR amplification using Pfu (Stratagene) followed by digestion via DpnI (Fermentas, Glen Burnie, MD, USA) to remove the templates. The mutagenic primer sequences were 5′-TCTGGCCCCGCGTTCTGTCGAATTTGCGCACCAA-3′ (XBP1s-Mut; m1) and 5′-TTCTGTCGGCCACGCGCATTTGCGGGGCAGGAT-3′ (XBP1s-Mut; m2). Mutations were subsequently sequenced, confirmed and used for promoter analysis.
Chromatin immunoprecipitation assay
Cells were differentiated for the indicated times, crosslinked with formaldehyde and sonicated to generate 200 bp per 1000 bp DNA fragments. The fragmented chromatin samples were immunoprecipitated with rabbit anti-mouse XBP1 (5 μg, Santa Cruz Biotechnology) and reverse crosslinked, purified and analyzed using real-time PCR according to the supplier’s protocol (Millipore, Billerica, MA, USA). Primers for the amplicons of the mouse miR-148a promoter region are listed in Table 1. DNA samples that were immunoprecipitated with rabbit IgG were used as negative controls.
Statistical analysis
All values are expressed as the mean±s.e.m. from at least three independent experiments. The data were analyzed using Student’s t-test or one- or two-way analysis of variance followed by Tukey’s multiple comparison test. Statistical analyses were performed using GraphPad Prism 5 for Windows (GraphPad Prism Software, La Jolla, CA, USA). In all cases, statistical significance was set at P<0.05.
Results
XBP1 regulates the stability of Wnt10b mRNA in 3T3-L1 cells
When we transfected siXBP1 into 3T3-L1 preadipocytes (Figure 1a), the Wnt10b mRNA level was significantly increased by up to 60% (P<0.05; Figure 1b). To examine the involvement of XBP1s in stabilizing Wnt10b mRNA, the rate of Wnt10b mRNA degradation was measured in 3T3-L1 cells treated with actinomycin D. Wnt10b mRNA was rapidly degraded, with a half-life of 3 h in siCON-transfected cells. However, the stability of Wnt10b mRNA was significantly increased by siXBP1 transfection, and it remained steady for 4 h (P<0.05; Figure 1c). These findings suggest that regulation of mRNA stability may be an additional mechanism involved in XBP1-induced downregulation of Wnt10b mRNA.

XBP1 regulates Wnt10b mRNA stability in 3T3-L1 preadipocytes. Cells were transfected for 24 h with 20 nM of control (siCON) or siXBP1. (a) Protein levels of XBP1s analyzed with immunoblot analysis. Beta-actin was used as a loading control. (b) mRNA levels of Wnt10b determined by real-time PCR. (c) Wnt10b mRNA stability determined by real-time PCR in cells treated with actinomycin D (5 μg ml−1) for the indicated times. The data are presented as percentages of Wnt10b mRNA relative to the level at 0 h (100%). All data are expressed as the mean±s.e.m. of three independent experiments (*P<0.05 compared with the siCON).
miR-148a suppresses Wnt/β-catenin signaling
To identify microRNAs that could bind to Wnt10b mRNA, we used computational prediction algorithms and identified a set of candidate microRNAs (data not shown). We selected miR-148a based on previous reports, indicating that Wnt10b mRNA may be a target of miR-148a.6, 22 We first examined the temporal pattern of miR-148a expression during adipogenesis. As shown in Supplementary Figure S1, miR-148a significantly increased on days 6 and 8 after induction. To verify the role of miR-148a in the regulation of Wnt10b mRNA, we examined the effect of a miR-148a mimic on Wnt10b mRNA levels in 3T3-L1 cells (Figure 2). When compared with a non-targeting control, transfection of the miR-148a mimic for 48 h increased intracellular miR-148a levels in 3T3-L1 cells (P<0.05; Figure 2a). In these cells, Wnt10b expression was significantly downregulated at both the mRNA and protein levels (Figure 2b), suggesting that Wnt10b mRNA was a potential target of miR-148a. A miR-148a-binding site at the 3′ untranslated repeat (UTR) of Wnt10b mRNA is evolutionarily conserved across mammals (Figure 2c).22 To confirm the importance of this sequence in 3T3-L1 cells, we conducted reporter analysis using a vector in which the 3′UTR of Wnt10b mRNA containing the miR-148a-binding motif was cloned downstream of EGFP (pEGFP-3′UTR). The results showed that the miR-148a mimic significantly suppressed GFP expression in 3T3-L1 cells transfected with pEGFP-3′UTR (P<0.05; Figure 2d). Moreover, Super TOPFlash assay results revealed that almost 50% of the β-catenin activity was reduced by the miR-148a mimic (P<0.05; Figure 2e). These findings indicate that miR-148a binds to Wnt10b mRNA and silences its expression in 3T3-L1 cells, thus suppressing the downstream β-catenin signaling pathway.

Wnt10b mRNA is a direct target of miR-148a in 3T3-L1 preadipocytes. Cells were transfected with 20 nM of siCON or miR-148a mimic for 24 h. (a) Expression levels of mature miR-148a were analyzed using real-time PCR. (b) Expression of Wnt10b mRNA and protein was analyzed using real-time PCR and immunoblot analyses. Immunoblot bands were quantified with densitometry analysis. Beta-actin was used as the loading control. (c) Schematic representation of Wnt10b 3′UTR with the putative miR-148a-binding site in mammals. Seed regions are shown with a gray background. (d) Green fluorescent protein (GFP) expression levels analyzed by immunoblot analysis and densitometry. Empty vector (pEGFP) or pEGFP reporter vector containing Wnt10b 3′UTR (pEGFP-3′UTR) was co-transfected with siCON or miR-148a mimic in HEK293T cells. Beta-actin served as the loading control. (e) Luciferase activities. Super TOPFlash construct was co-transfected with siCON or miR-148a mimic for 48 h. Beta-catenin was used as a positive control. The data are expressed as the mean±s.e.m. from three separate experiments (*P<0.05 compared with the siCON).
XBP1 directly regulates miR-148a transcription
Next, we examined the connection between XBP1s and miR-148a in 3T3-L1 preadipocytes by silencing or overexpressing XBP1s. As shown in Figure 3a, miR-148a levels were positively correlated with those of XBP1s. The level of mature form of miR-148a was downregulated (~75%) by siXBP1 (P<0.05; Figure 3a) but upregulated (~60%) by XBP1 overexpression (P<0.05; Figure 3b). The primary miR-148a transcript level was assessed in wild type or XBP1-KD cells to determine whether XBP1s could regulate miR-148a transcription. The results from PCR using primer sets designed to cover two different putative regions of miR-148a precursor sequences (Figure 3c, left) clearly showed that amplicons in XBP1-KD cells were significantly smaller than those in wild-type cells (P<0.05; Figure 3c, right), suggesting that XBP1s directly regulates miR-148a transcription.

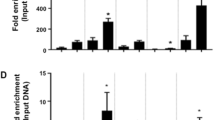
miR-148a transcription is directly regulated by XBP1s in 3T3-L1 preadipocytes. (a, b) Expression levels of XBP1s mRNA and miR-148a measured with real-time PCR. Cells were transfected for 24 h with siCON or siXBP1 (a) and empty vector (EV) or mouse pcDNA3.1-mXBP1s (b). (c) Schematic chromosomal position of the mouse miR-148a gene. miR-148a precursor sequences are shown with a gray background. The levels of primary miR-148a (Pri-miR-148a) were measured with conventional RT-PCR analysis and densitometry. (d) Putative Pri-miR-148a transcription start sites (TSSs) were predicted as described in the Materials and Methods section, and deletion luciferase reporter constructs (#1 to #6) were generated as indicated. (e, f) Luciferase activities. Each deletion construct (e) or construct #3 (f) were co-transfected into HEK293T cells with EV or pcDNA3.1-mXBP1s as indicated, and luciferase activities were determined from each cell lysate. (g, h) DNA sequences of two putative XBP1-binding sites in the miR-148a promoter region are shown in gray. To determine whether these sites are responsible for XBP1 binding, two different mutants (m1 and m2) were created using site-directed mutagenesis as indicated. Then, miR-148a promoter activities were measured in cells co-transfected with wild type (WT) or each mutant construct and pcDNA3.1-mXBP1s. (i) Chromatin immunoprecipitation analysis performed with anti-XBP1 antibody or control IgG using lysates from 3T3-L1 preadipocytes. The amount of immunoprecipitated miR-148a promoter region (−450/−302 bp) was quantified with PCR. For input, total chromatin (1/10 volume of chromatin used for immunoprecipitation) was subjected to PCR. All data are presented as the mean±s.e.m. of three independent experiments (*P<0.05 compared with siCON, EV or WT).
To identify the binding site(s) of XBP1s in the promoter of miR-148a, we identified a transcription start site (TSS) in the upstream region of the miR-148a gene using cap analysis of gene expression data (http://fantom.gsc.riken.jp/4/gev/gbrowse/mm9/; Supplementary Figure S2). Six luciferase reporter constructs were created by cloning DNA fragments that were sequentially deleted up to 1.5 kb upstream of the TSS (Figure 3d). The functional activity of each deletion mutant was then determined. As shown in Figure 3e, the luciferase activity was the highest (12-fold, P<0.05) in the construct containing the 0.5-kb DNA fragment from TSS (#3), and it was dose-dependently increased according to the amount of XBP1 expression vector (P<0.05; Figure 3f). However, no reporter activity was found in the construct containing a 0.3-kb fragment from TSS, suggesting that the XBP1 response elements (REs) are located between 0.3 and 0.5 kb upstream of TSS of miR-148a gene. On the basis of previous studies,23 we located two potential XBP1s REs at −301~−296 and −311~−307 (Figure 3g). To identify whether these REs are functional, we introduced site-directed mutations into each candidate RE (Figure 3g) and performed promoter analysis. When we co-transfected these reporters with XBP1s into 3T3-L1 cells, miR-148a promoter activity was reduced by 50% only in the construct containing mutations in the more upstream RE (Figure 3h). Chromatin immunoprecipitation analysis also revealed that XBP1s could directly bind to the miR-148a promoter region containing this motif (Figure 3i). Taken together, these results clearly demonstrate that XBP1 is a novel regulatory factor for miR-148a transcription through direct binding to an XBP1 RE at the promoter region of miR-148a in 3T3-L1 cells.
miR-148a mimic restores adipogenic potential in XBP1-deficient cells
We previously reported that XBP1 is an important regulator of Wnt10b during adipogenesis.16 Here we provide a novel mechanism for XBP1-mediated suppression of Wnt10b through directly inducing miR-148a. To verify that miR-148a is a downstream functional effector in XBP1-induced adipogenesis, we examined whether a miR-148a mimic could reverse Wnt10b mRNA levels and therefore rescue adipogenesis in XBP1-deficient 3T3-L1 cells. As shown in Figure 1, Wnt10b mRNA was increased by XBP1 knockdown, and it was significantly (P<0.05) suppressed by the miR-148a mimic (Figure 4a). However, results from promoter analysis revealed that miR-148a failed to affect Wnt10b transcription (Figure 4b). These findings clearly suggest that there are two distinct mechanisms involved in XBP1-mediated Wnt10b regulation: direct transcriptional repression and miR-148a-mediated post-transcriptional silencing.

Ectopic expression of miR-148a rescues adipogenesis in XBP1-deficient 3T3-L1 preadipocytes. (a, b) Cells were transfected with siCON, miR-148a mimic and siXBP1 as indicated. (a) miR-148a and Wnt10b mRNA levels were determined with real-time PCR. (b) Luciferase activities. Cells were co-transfected with pGL2-mWnt10b promoter-Luc reporter vector, and promoter activity was assessed at 48 h post transfection. (c–g) Determination of adipogenesis. siCON or miR-148a was transfected into 3T3-L1 cells in which XBP1 is stably knocked down, and adipogenic differentiation was induced for 6 days. (c) miR-148a and Wnt10b mRNA levels measured with real-time PCR. (d, e) Levels of intracellular triacylglycerol accumulation assessed by Nile Red staining (d) and measurement of triacylglycerol content (e). (f, g) Expression of PPARγ2, adiponectin and C/EBPα examined by real-time PCR (f) and immunoblot analyses (g). The data are expressed as the mean±s.e.m. of representative analyses from three separate experiments (*P<0.05 compared with the siCON; #P<0.05 compared with the miR-148a mimic-untreated; NS, no significant difference).
As expected, the miR-148a mimic significantly decreased Wnt10b mRNA levels at 6 days post differentiation (P<0.05; Figure 4c) and increased Nile Red staining (threefold) and intracellular triacylglyceride accumulation (30%; P<0.05; Figures 4d and e). These findings strongly indicated that the adipogenic potential of XBP1-KD cells could be successfully recovered by miR-148a overexpression. An adipogenic key transcription factor (PPARγ) and a mature adipocyte marker (adiponectin) were also significantly increased by miR-148a mimic treatment at both the mRNA and protein levels (P<0.05; Figures 4f and g). However, C/EBPα levels showed a tendency to increase by miR-148a mimic; however, this was not statistically significant. These data demonstrate that miR-148a is an important downstream effector molecule of XBP1-induced adipogenesis by silencing Wnt10b mRNA.
Discussion
miR-148a is highly abundant in adipose tissues.24, 25 It is strongly induced during adipogenic differentiation in mouse 3T3-L1 cells6, 26 and adipose-derived mesenchymal stem cells.7, 27 Moreover, miR-148a levels are increased in obese mice and humans.7 These findings strongly suggest an important role for miR-148a in the regulation of adipogenesis and in the development of obesity or other metabolic disorders. However, the precise regulatory mechanisms involved in its transcription remain unclear. From that point of view, the most notable finding in our study is that XBP1s may be a novel transcriptional activator for miR-148a in differentiating 3T3-L1 cells. When we altered the expression of XBP1 in 3T3-L1 cells, miR-148a levels changed accordingly. The levels of miR-148a were significantly downregulated by XBP1 knockdown during adipogenic differentiation (Supplementary Figure S1). In addition, we revealed an XBP1-specific RE in the promoter region of miR-148a. These findings clearly suggest that XBP1 is an upstream regulator of miR-148a transcription. However, when we compared expression patterns in the early differentiation period, no correlation was found between XBP1s and miR-148a (Supplementary Figure S1). As we previously reported, the level of XBP1 mRNA was increased in two phases: a strong peak at ~6 h and a less potent peak on day 6 after the induction.13 miR-148a expression, consistent with previous reports, was not increased within 24 h, but it began to increase at 6 days after adipogenic induction (Supplementary Figure S1).7, 26 In the same context, the cAMP-response element-binding protein (CREB), which is known to regulate adipogenesis during the very early period, has been recently suggested as a putative upstream regulator of miR-148a.7, 28 Therefore, it is reasonable to assume that additional transcriptional factors or cofactors induced by adipogenic differentiation stimuli may be needed for XBP1- or CREB mediated miR-148a production.
The Wnt/β-catenin pathway has been implicated in many pathophysiological aspects of adipose tissue metabolism and disorders. Therefore, manipulating the Wnt/β-catenin pathway in adipose tissue is an attractive drug-development strategy to combat obesity-associated metabolic complications. Of a total of 19 Wnts, Wnt1 and Wnt10b are regarded as typical anti-adipogenic isotypes,29 and the latter is considered to be the most potent anti-adipogenic Wnt ligand. Previously, we suggested that Wnt10b transcription is specifically repressed by XBP1s during adipogenic differentiation of 3T3-L1 cells,16 and, in this study, we showed the involvement of miR-148a in XBP1-mediated suppression of Wnt10b. A miR-148a mimic significantly downregulated Wnt10b at both the mRNA and protein levels by binding to the 3′UTR of Wnt10b mRNA in 3T3-L1 cells. Consistent with our findings, Aprelikova et al.22 recently suggested that Wnt10b is a potential target of miR-148a in cancer-associated fibroblasts. These findings provide novel information that XBP1s can downregulate Wnt10b expression through both transcriptional and miR-148a-mediated post-transcriptional mechanisms to facilitate adipogenesis of 3T3-L1 cells. However, the evidence provided in this work is not sufficient to determine the relative contribution of direct and indirect mechanisms to XBP1-induced Wnt10b repression. We may be able to determine such contributions by comparing the temporal expression patterns of XBP1s, Wnt10b and miR-148a during adipocyte differentiation. As mentioned earlier, the XBP1 mRNA level was increased in both the early and late phases, whereas miR-148a was increased only in the late phase of adipogenesis (Supplementary Figure S1). On the basis of these findings, the simplest explanation for this discrepancy in temporal expression patterns between XBP1s and miR-148a is as follows: XBP1 downregulates Wnt10b mRNA by a transcriptional mechanism during the early phase16 and by miR-148a-mediated post-transcriptional mechanism in the late phase of adipogenesis. Moreover, a miR-148a inhibitor increased Wnt10b mRNA levels only in the late phase after induction (Supplementary Figure S3), suggesting that miR-148a may have no effect on Wnt10b mRNA in the early phase. Further research will be needed to reveal the mechanisms involved in the time-dependent and interactive regulation among miR-148a, Wnt10b and XBP1s during adipogenesis.
Wnt10b inhibits adipocyte differentiation by blocking the induction of C/EBPα and PPARγ in the early phase of adipogenesis;30, 31 however, its role in the late phase is relatively unknown. What is the role of miR-148a-induced silencing of Wnt10b in the late phase of adipogenic differentiation? In this study, the miR-148a mimic downregulated the mRNA level of Wnt10b, and it concurrently rescued adipogenic potential and the expression of PPARγ2 in XBP1-KD cells (Figure 4). Therefore, it is plausible that in addition to directly suppressing Wnt10b transcription during the early phase of adipogenesis, XBP1s may intensify adipogenic differentiation by inducing miR-148a, which, in turn, can disinhibit Wnt10b-mediated suppression of PPARγ2 during the late phase of adipogenesis. Moreover, considering that important pro- and anti-adipogenic molecules such as PPARγ2,13 Wnt10b16 and C/EBPα14 are regulated by XBP1s, our data identifying miR-148a as an additional target molecule for XBP1s strongly support the idea that XBP1 plays a critical role in adipogenesis through multiple mechanisms (Figure 5).

Diagram of XBP1-mediated adipogenesis. Accumulating evidence has indicated that XBP1 plays a critical role in adipogenesis by regulating important pro- and anti-adipogenic molecules such as PPARγ2, Wnt10b and C/EBPα. In this study, we demonstrate that miR-148a is a novel downstream effector in XBP1-mediated adipogenesis through silencing Wnt10b mRNA. Refer to the Discussion section for a detailed explanation. ACL, ATP-citrate lyase; DNMT1, DNA methyltransferase 1; XBP1, X-box binding protein 1.
It is well known that each microRNA can regulate the expression of more than 100 target genes.32 Thus, it is not unlikely that molecular targets other than Wnt10b may be involved in XBP1-mediated adipogenesis. For example, DNA methyltransferase 1 was reported to be a target of miR-148a during adipogenesis,26 indicating that miR-148a might participate in XBP1-induced adipogenesis through Wnt-independent mechanisms (Figure 5). In addition, while we were preparing this manuscript, Wnt1 was suggested as a potential target of miR-148a in human adipose-derived mesenchymal stem cells.7 One notable finding from this study is that, in contrast to our findings, the Wnt10b level was not reduced by miR-148a. In this context, the transcription of only Wnt10b, but not Wnt1, was found to be suppressed by XBP1s in our previous report.16 With the information currently available, we could not explain the differential regulatory mechanisms involved in miR-148a-induced silencing of Wnt1 and Wnt10b. However, part of the answer to this question may be found in several lines of evidence, suggesting that microRNA functions are frequently cell-type-specific. For example, many studies have reported that the inhibitory effects of miR-148a on Wnt10b depend on the cellular context.22, 33, 34 It has also been reported that miR-148a silences DNA methyltransferase 1 in 3T3-L1 cells26 but not in human mesenchymal stromal cells,7 suggesting the presence of species-dependent activities of miR-148a. Further evidence is required to determine whether miR-148a activities are cell-type- or species-specific.
In summary, for the first time, we found that XBP1s could stimulate adipocyte differentiation by directly activating miR-148a-mediated suppression of Wnt10b. With the known regulatory function of XBP1s on the key pro- and anti-adipogenic molecules such as C/EBPα, PPARγ2 and Wnt10b, our data suggesting a novel mechanism involving miR-148a also support our hypothesis that XBP1 may be an essential regulator for adipogenesis.
References
Gustafson B, Hedjazifar S, Gogg S, Hammarstedt A, Smith U . Insulin resistance and impaired adipogenesis. Trends Endocrinol Metab 2015; 26: 193–200.
Longo KA, Wright WS, Kang S, Gerin I, Chiang SH, Lucas PC et al. Wnt10b inhibits development of white and brown adipose tissues. J Biol Chem 2004; 279: 35503–35509.
Wright WS, Longo KA, Dolinsky VW, Gerin I, Kang S, Bennett CN et al. Wnt10b inhibits obesity in ob/ob and agouti mice. Diabetes 2007; 56: 295–303.
Chen L, Song J, Cui J, Hou J, Zheng X, Li C et al. MicroRNAs regulate adipocyte differentiation. Cell Biol Int 2013; 37: 533–546.
Zhang R, Wang D, Xia Z, Chen C, Cheng P, Xie H et al. The role of microRNAs in adipocyte differentiation. Front Med 2013; 7: 223–230.
Qin L, Chen Y, Niu Y, Chen W, Wang Q, Xiao S et al. A deep investigation into the adipogenesis mechanism: profile of microRNAs regulating adipogenesis by modulating the canonical Wnt/beta-catenin signaling pathway. BMC Genomics 2010; 11: 320.
Shi C, Zhang M, Tong M, Yang L, Pang L, Chen L et al. miR-148a is associated with obesity and modulates adipocyte differentiation of mesenchymal stem cells through Wnt signaling. Sci Rep 2015; 5: 9930.
Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001; 412: 300–307.
Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008; 134: 743–756.
Lee AH, Chu GC, Iwakoshi NN, Glimcher LH . XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J 2005; 24: 4368–4380.
Iwakoshi NN, Pypaert M, Glimcher LH . The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med 2007; 204: 2267–2275.
Bettigole SE, Lis R, Adoro S, Lee AH, Spencer LA, Weller PF et al. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat Immunol 2015; 16: 829–837.
Cho YM, Kwak SN, Joo NS, Kim DH, Lee AH, Kim KS et al. X-box binding protein 1 is a novel key regulator of peroxisome proliferator-activated receptor gamma2. FEBS J 2014; 281: 5132–5146.
Sha H, He Y, Chen H, Wang C, Zenno A, Shi H et al. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab 2009; 9: 556–564.
Basseri S, Lhotak S, Sharma AM, Austin RC . The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J Lipid Res 2009; 50: 2486–2501.
Cho YM, Kim DH, Kwak SN, Jeong SW, Kwon OJ . X-box binding protein 1 enhances adipogenic differentiation of 3T3-L1 cells through the downregulation of Wnt10b expression. FEBS Lett 2013; 587: 1644–1649.
Helledie T, Jorgensen C, Antonius M, Krogsdam AM, Kratchmarova I, Kristiansen K et al. Role of adipocyte lipid-binding protein (ALBP) and acyl-coA binding protein (ACBP) in PPAR-mediated transactivation. Mol Cell Biochem 2002; 239: 157–164.
Seo JB, Choe SS, Jeong HW, Park SW, Shin HJ, Choi SM et al. Anti-obesity effects of Lysimachia foenum-graecum characterized by decreased adipogenesis and regulated lipid metabolism. Exp Mol Med 2011; 43: 205–215.
Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH . Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci USA 2003; 100: 9946–9951.
Lee AH, Iwakoshi NN, Glimcher LH . XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 2003; 23: 7448–7459.
Park K, Lee K, Zhang B, Zhou T, He X, Gao G et al. Identification of a novel inhibitor of the canonical Wnt pathway. Mol Cell Biol 2011; 31: 3038–3051.
Aprelikova O, Palla J, Hibler B, Yu X, Greer YE, Yi M et al. Silencing of miR-148a in cancer-associated fibroblasts results in WNT10B-mediated stimulation of tumor cell motility. Oncogene 2013; 32: 3246–3253.
Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C et al. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell 2007; 27: 53–66.
Ma J, Jiang Z, He S, Liu Y, Chen L, Long K et al. Intrinsic features in microRNA transcriptomes link porcine visceral rather than subcutaneous adipose tissues to metabolic risk. PLoS ONE 2013; 8: e80041.
Li G, Li Y, Li X, Ning X, Li M, Yang G . MicroRNA identity and abundance in developing swine adipose tissue as determined by Solexa sequencing. J Cell Biochem 2011; 112: 1318–1328.
Londono Gentile T, Lu C, Lodato PM, Tse S, Olejniczak SH, Witze ES et al. DNMT1 is regulated by ATP-citrate lyase and maintains methylation patterns during adipocyte differentiation. Mol Cell Biol 2013; 33: 3864–3878.
Bengestrate L, Virtue S, Campbell M, Vidal-Puig A, Hadaschik D, Hahn P et al. Genome-wide profiling of microRNAs in adipose mesenchymal stem cell differentiation and mouse models of obesity. PLoS ONE 2011; 6: e21305.
Reusch JE, Colton LA, Klemm DJ . CREB activation induces adipogenesis in 3T3-L1 cells. Mol Cell Biol 2000; 20: 1008–1020.
Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL et al. Inhibition of adipogenesis by Wnt signaling. Science 2000; 289: 950–953.
Cawthorn WP, Bree AJ, Yao Y, Du B, Hemati N, Martinez-Santibanez G et al. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a beta-catenin-dependent mechanism. Bone 2012; 50: 477–489.
Lee H, Bae S, Yoon Y . Anti-adipogenic effects of 1,25-dihydroxyvitamin D3 are mediated by the maintenance of the wingless-type MMTV integration site/beta-catenin pathway. Int J Mol Med 2012; 30: 1219–1224.
Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ et al. Combinatorial microRNA target predictions. Nat Genet 2005; 37: 495–500.
Zheng B, Liang L, Wang C, Huang S, Cao X, Zha R et al. MicroRNA-148a suppresses tumor cell invasion and metastasis by downregulating ROCK1 in gastric cancer. Clin Cancer Res 2011; 17: 7574–7583.
Gokhale A, Kunder R, Goel A, Sarin R, Moiyadi A, Shenoy A et al. Distinctive microRNA signature of medulloblastomas associated with the WNT signaling pathway. J Cancer Res Ther 2010; 6: 521–529.
Acknowledgements
This research was supported by a grant (14172MFDS974) from the Ministry of Food and Drug Safety in 2015. We thank C Kim (Department of Biochemistry, College of Medicine, The Catholic University of Korea, Seoul, Korea) for the EGFP reporter construction. We also thank Professor Eun Kyung Lee (The Catholic University of Korea, Seoul, Korea) for helpful discussions and critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Cho, Y., Kim, TM., Hun Kim, D. et al. miR-148a is a downstream effector of X-box-binding protein 1 that silences Wnt10b during adipogenesis of 3T3-L1 cells. Exp Mol Med 48, e226 (2016). https://doi.org/10.1038/emm.2016.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2016.3

{kind=link}
{kind=link}
{kind=link}