
Abstract
The introduction of whole-exome sequencing into the Pediatric Genetics clinic has increased the identification of novel genes associated with neurodevelopmental disorders and congenital anomalies. This agnostic approach has shed light on multiple proteins and pathways not previously known to be associated with disease. Here we report eight subjects from six families with predicted loss of function variants in ZNF462, a zinc-finger protein of unknown function. These individuals have overlapping phenotypes that include ptosis, metopic ridging, craniosynostosis, dysgenesis of the corpus callosum, and developmental delay. We propose that ZNF462 plays an important role in embryonic development, and is associated with craniofacial and neurodevelopmental abnormalities.
Similar content being viewed by others


Introduction
The implementation of whole-exome sequencing (WES) in Pediatric Genetics clinics resulted in increased diagnosis rates as well as novel gene discovery.1 In some instances, the utilization of WES and gene-matching tools helped identify new Mendelian syndromes through reverse phenotyping.2 This is accomplished by in depth clinical evaluations of individuals with similar genetic variants, and emphasizes the importance of the dysmorphology exam in the genomic era.
Premature fusion of the metopic suture is associated with trigonocephaly or a triangular-shaped forehead when viewed from above. The classic diagnostic triad consists of forehead narrowing, biparietal widening, and pseudohypotelorism, but a CT scan is considered the gold standard for diagnosis.3 The majority of cases are isolated with an overall good prognosis; however,4, 5 there are several well-defined monogenic syndromes6, 7, 8 and chromosomal aberrations9, 10 associated with metopic craniosynostosis. In many of these conditions craniosynostosis is a common but not universal finding, and in some patients, only a metopic ridge without true trigonocephaly is observed.11 We performed WES in a four-generation family with significant metopic ridging and ptosis. We identified a unique nonsense variant in ZNF462 segregating among affected family members. Utilizing GeneMatcher12 and Decipher,13 we identified five additional unrelated individuals with de novo predicted loss-of-function (LOF) variants (including ZNF462 microdeletions) with an overlapping phenotype. Additional clinical findings included dysgenesis of the corpus callosum, dysmorphic features, and developmental delay with or without autism. We propose that haploinsufficiency of ZNF462 is associated with craniofacial anomalies, dysgenesis of corpus callosum, and developmental delay with variable expressivity.
Materials and methods
WES was performed for families 1–3. For family 1, sequencing was done at the NIH Intramural Sequencing Center (NISC) using the SeqCap EZ Exome version 3.0 capture kit (Roche NimbleGen, Madison, MI, USA) and the Illumina HiSeq2500 platform (Illumina, San Diego, CA, USA). Sequencing data were aligned to the human reference genome using Novoalign (Novocraft Technologies, Selangor, Malaysia). Variants were called using the in-house MPG caller. Detected variants were annotated and filtered using an in-house bioinformatics pipeline. We filtered for dominantly inherited heterozygous variants segregating among affected family members with a mean allele frequency of <0.001 in the Exome Aggregation Consortium (ExAC) database. For families 2 and 3, clinical exome sequencing was performed at GeneDx (Gaithersburg, MD, USA). Sanger confirmation of the variants detected by WES was performed in all cases (Supplementary Figure 1). The proband and parents in family 4 underwent a chromosomal microarray SurePrint G3 4 × 180 k (Agilent Technologies, Santa Clara, CA, USA). The proband and parents in family 5 underwent a chromosomal microarray CytoSure 180 k (Oxford Gene Technology, Oxfordshire, UK). The proband and parents from family 6 participated in the Deciphering Developmental Disorders (DDD) project, a UK-based WES study.14 All families were consented for genetic testing and publication of clinical and sequencing findings.
Results
Case descriptions
The proband in family 1 (Figure 1, pedigree 1, subject IV1) is a 2-year-old female born at 39 weeks gestation. A fetal anomaly ultrasound (US) at 18 weeks detected agenesis of the corpus callosum (ACC) with mild ventriculomegaly. Physical examination after birth was normal except for a pointed forehead. At 3 months a brain MRI showed ACC with associated colpocephaly. A CT scan did not meet criteria for metopic craniosynostosis and surgery was not indicated (Supplementary Figure 2). She had normal growth and development at 2 years of age. Physical examination findings included: a pointed forehead, bilateral ptosis, arched eyebrows, synophrys, a small upturned nose, long philtrum, and a thin upper lip. (Table 1; Figure 2a). The proband has a positive family history of metopic ridging, which affects her elder sister (IV2), father (III2), and paternal great grandfather (I1). The proband’s sister was 4 years old during her last evaluation and demonstrated normal growth and development. Her physical exam was significant for a pointed forehead, ptosis, down-slanted palpebral fissures, and a congenital capillary malformation (port wine stain) involving the right side of her neck, shoulder, and upper left arm. A fetal anomaly scan during the pregnancy was normal without ACC. The father had a pointed forehead and mild left ptosis, but no other dysmorphic features. The paternal grandmother had bilateral ptosis, without metopic ridging or dysmorphic features. The paternal great grandfather was not examined but was reported to have a pointed forehead. None of the proband’s family members had brain imaging or surgery for craniosynostosis.

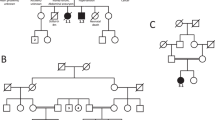
The pedigrees of six families with ZNF462 variants.

Facial photographs of subjects with ZNF462 variants. (a) From left to right: proband (6 months), a bird’s eye view of her head, sister (1 month) with a bird’s eye view of her head and the father from family 1. (b) Proband from family 2 at the age of 2 years. (c) Proband from family 3 at the age of 2 years. (d) Proband from family 4 at the age of 3 and 5 years. (e) Proband from family 5 at the age of 4 years. (f) Proband from family 6 at 7 years. Note: more recent photos showing more significant ptosis in both sisters from family 1 were not approved for publication by the family.
The proband in family 2 (Figure 1, pedigree 2) is a 2-year-old male, born full term after an uneventful pregnancy and delivery. At 2 years he was diagnosed with expressive speech delay and autistic spectrum disorder (ASD). On physical exam (Figure 2b) he had a pointed forehead, down slanting palpebral fissures, arched eyebrows, small widely spaced teeth, asymmetric ears, small upturned nose with a short philtrum and a small columella, fifth finger clinodactyly, bilateral single transverse palmar crease, and mild 2–3 toe syndactyly. Brain MRI was normal. Metopic ridging with a triangular-shaped head was diagnosed clinically, and neither a head CT nor surgical intervention was performed. Previous genetic testing included normal Fragile X testing and array CGH.
The proband in family 3 (Figure 1, pedigree 3) is a 32-month-old male born at 37 weeks via spontaneous vaginal delivery. The pregnancy was uneventful and the delivery was uncomplicated. On routine pulse oximetry screening, he was found to be hypoxemic and an echocardiogram revealed a complete D-transposition of the great vessels with intact ventricular septum, and a large patent ductus arteriosus. A head US and brain MRI showed a hypoplastic corpus callosum with mild ventriculomegaly (Supplementary Figure 2). At 4 weeks of age, he underwent gastrostomy tube placement and Nissen fundoplication due to silent aspiration confirmed by an abnormal swallow study. Subsequently he developed progressive right-sided plagiocephaly. A head CT at 7 months demonstrated a unilateral right lambdoid synostosis (Supplementary Figure 2). Surgical release of the lambdoid synostosis was performed at 12 months. On physical exam (Figure 2c), there was metopic ridging, mild down slanting palpebral fissures with left ptosis, and a bulbous nasal tip. The ears were protuberant, with the right ear over folded superiorly, crumpled laterally, and posteriorly rotated and the left simple and cupped. The superior labial frenulum was broad and there was mild nail hypoplasia. Motor and speech delays were noted on clinical evaluation at 19 and 32 months. He sat supported at 8 months and, pulled to a stand at 19 months. At 32 months he had 10 words and several signs, and was noted to have unusual hand flapping when excited, looked at individuals from the sides of his eyes and did not demonstrate joint attention or reciprocal play. Previous genetic testing included normal Fragile X and array CGH.
The proband in family 4 (Figure 1, pedigree 4) is the first child of unrelated Norwegian parents. She was born full term. Due to hypotonia and bilateral ptosis in the newborn period, she underwent a brain MRI that demonstrated hypoplasia of the corpus callosum (Supplementary Figure 2) with a thin anterior part and a missing posterior part. She had poor weight gain in the first few weeks that later on resolved. She walked at 15 months and speech development was normal. On physical exam (Figure 2d) she has bilateral ptosis, left strabismus and epicanthal folds, arched eyebrows, and a short philtrum. At the age of 5 years, she attended kindergarten and there was no concern about her development.
The proband in family 5 (Figure 1, pedigree 5) is a 15-year-old female of Swedish descent and was born full term. She presented with a ventricular septal defect, and fine and gross motor delay. Later she was diagnosed with mild intellectual disability, autism spectrum disorder, attention deficit disorder, and obsessive compulsive disorder. She had an increased appetite and developed overweight in her teenage years. On physical exam (Figure 2e) she had malformed and discolored permanent teeth, marked cupid bow, broad upper lip frenulum, and a broad face. Brain imaging was not performed and there was no history of craniosynostosis.
The proband in family 6 (Figure 1, pedigree 6) is a 9-year-old male. He was born full term after an uneventful pregnancy and delivery. He was described by his mother as a floppy baby. He had surgery for an umbilical hernia at 9 months. He sat independently at 10 months, took his first steps by 16 months and was walking by 18 months. He demonstrated joint laxity and easy bruising from a young age. He suffered from frequent falls although by 3 years, he was able to climb up stairs, attempting to jump, and falling less frequently. In addition, he had fine motor delay and speech delay. He wore glasses for hypermetropia. At the age of 7 years, he had four episodes that were investigated as possible absence seizures. EEG and brain MRI scan were normal. On physical exam (Figure 2f), he had prominent ears with thickened helices and a slightly myopathic facies. He has down slanting palpebral fissures and a mild ptosis bilaterally. He was not diagnosed with craniosynostosis. However, he had some facial asymmetry and history of coronal suture ridging. Previous genetic testing included normal Fragile X and array CGH.
Sequencing and microarray results
In family 1, the nonsense variant c.3787C>T p.(Arg1263*) in ZNF462 (NM_021224.4) segregated in four family members (IV1, IV2, III2, and II2); I1 was not tested. There were no rare variants in genes previously known to be associated with craniosynostosis or ACC. In families 2, 3, and 6, the probands underwent trio-based WES, which revealed ZNF462 de novo frameshift variants (c.2979_2980delinsA p.(Val994Trpfs*147), c.4263delA p.(Glu1422Serfs*6), and c.5145delC p.(Tyr1716Thrfs*28)), respectively. For the proband in family 3, a paternally inherited variant of uncertain significance (VUS) in FOXP2 (c.776-5T>G, NM_014491.3) and a mitochondrial VUS (m.14787T>C p.(I14T), NC_012920.1) in the MT-CYB gene at 30% heteroplasmy was found. No additional VUS was reported for the proband in family 2 and 6. All four sequence variants were located within exon 3 of ZNF462 (Figure 3a) and are listed in http://ZNF462.LOVD.nl IDs 96028-30, 100323. The proband in family 4 underwent chromosomal microarray, which identified a de novo microdeletion at 9q31.2 encompassing the entire ZNF462 gene and two additional OMIM genes not known to be associated with disease: RAD23B (MIM: 600062), KLF4 (MIM: 602253) (Figure 3b; Supplementary Figure 3). The proband in family 5 had a larger de novo microdeletion that included a part of TMEM38B (MIM: 611236) as well (Figure 3b). This gene was described in autosomal recessive osteogenesis imperfecta type XIV and haploinsufficiency is not known to be associated with the disease. Proband 5 had in addition a paternally inherited 374 kb duplication of uncertain significance on 6q22.31, Chr6:g(118697303-119071611)dup (hg19).

ZNF462 variants and microdeletions. (a) A diagram showing the ZNF462 coding exons and introns (NM_021224.4). Note all detected variants are in the third exon and therefore likely result in nonsense-mediated mRNA decay. Chromatograms are shown in Supplementary Figure 1. (b) 9q31.2 microdeletions detected in the probands from families 4 and 5. Array data are shown in Supplementary Figure 3.
Discussion
We describe eight subjects with predicted LOF variants in ZNF462 and overlapping clinical findings as summarized in Table 1. Shared features include metopic ridging or lambdoid craniosynostosis (5/8), dysgenesis of the corpus callosum (3/8), ptosis (7/8), and developmental delay with or without autistic features (4/8). In addition, we identified overlapping dysmorphic features in most subjects such as arched eyebrows, down slanting palpebral fissures, epicanthal folds, wide philtrum, and a short upturned nose with a bulbous tip (Table 1; Figure 2). In order to capture all ZNF462 LOF variants, we searched the Decipher database and found 12 reports of individuals with ZNF462 deletions. All deletions were reported to be de novo or of unknown inheritance. Here, we included detailed clinical information of two individuals with 1.6 and 1.9 MB deletions (Decipher IDs 261011, 274871).13 The other deletions are larger than 4.8 MB and include dozens of genes. It is therefore difficult to determine the effect of ZNF462 in those cases and blended phenotypes are possible. In review of the literature there is a report of a de novo translocation, t(2;9)(p24;q32), disrupting ZNF462 and ASXL2 (MIM: 612991).15 That individual had a similar phenotype to participants in our study, consisting of ACC, ptosis, and developmental delay.16 There were additional congenital anomalies not reported here and more severe intellectual disability, likely the result of a blended phenotype secondary to both ZNF462 and ASXL2 haploinsufficiency.17 The significant clinical overlap of the eight individuals we report and an additional case from the literature strongly support a similar etiology.
In family 1, the phenotype associated with ZNF462 LOF variants was variable and ranged between isolated ptosis in subject II2 and significant dysmorphisms with ACC in the proband (IV1). Variable expressivity has been previously reported in syndromic craniosynostosis18, 19 and for genes involved in brain development.20, 21, 22 This may result from environmental or other genetic factors contributing to the phenotype. Here, it is important to acknowledge the possibility of a mild or unrecognized phenotype in a parent and the importance of testing parents for the variant. As found in three individuals in our cohort, ASD disorder is an important component of the ZNF462-associated phenotype. Interestingly, de novo missense and truncating variants in ZNF462 were reported in individuals with ASD23, 24, 25 and ZNF462 is listed in Sfari Gene database.26
ZNF462, also known as KIAA1803, encodes a C2H2-type zinc-finger protein of unknown function. Expressed in the nuclear compartment, gene ontology annotations include transcriptional regulation and chromatin organization. The ZNF462 sequence is highly conserved in most mammals, but not well conserved in non-vertebrates. Also, we know that ZNF462 is intolerant of LOF variants (ExAC pLi score of 1.0),27 and was listed as one of the top 1000 genes significantly lacking functional coding variation, suggesting it is likely associated with disease.28 According to the Mouse Genome Informatics database, broad expression is seen in mouse embryos with high levels in the embryonic ectoderm, head, and brain. Four of the subjects we describe have truncating variants in ZNF462, which are predicted to result in loss of function due to nonsense-mediated mRNA decay. Two other participants had a deletion of this gene, supporting haploinsufficiency as the mechanism of pathogenicity. While further functional studies are necessary, the clinical findings reported here suggest ZNF462 might be involved in transcriptional regulation during embryogenesis affecting neural crest migration and brain development.
In conclusion, we propose that haploinsufficiency of ZNF462 results in craniofacial anomalies and developmental delay with variable expressivity. Beyond the clinical implications, we shed light on an under-characterized protein likely involved in craniofacial and brain development.
References
Sawyer SL, Hartley T, Dyment DA et al: Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin Genet 2016; 89: 275–284.
Uliana V, Percesepe A : Reverse phenotyping comes of age. Mol Genet Metab 2016; 118: 230–231.
Birgfeld CB, Saltzman BS, Hing AV et al: Making the diagnosis: metopic ridge versus metopic craniosynostosis. J Craniofac Surg 2013; 24: 178–185.
Kini U, Hurst JA, Byren JC, Wall SA, Johnson D, Wilkie AO : Etiological heterogeneity and clinical characteristics of metopic synostosis: evidence from a tertiary craniofacial unit. Am J Med Genet A 2010; 152A: 1383–1389.
Lajeunie E, Le Merrer M, Marchac D, Renier D : Syndromal and nonsyndromal primary trigonocephaly: analysis of a series of 237 patients. Am J Med Genet 1998; 75: 211–215.
Sargent C, Burn J, Baraitser M, Pembrey ME : Trigonocephaly and the Opitz C syndrome. J Med Genet 1985; 22: 39–45.
Nakane T, Kubota T, Fukushima Y, Hata Y, Ishii J, Komiyama A : Opitz trigonocephaly (C)-like syndrome, or Bohring-Opitz syndrome: another example. Am J Med Genet 2000; 92: 361–362.
Salinas-Torres VM : Say-Meyer syndrome: additional manifestations in a new patient and phenotypic assessment. Childs Nerv Syst 2015; 31: 1181–1187.
Mattina T, Perrotta CS, Grossfeld P : Jacobsen syndrome. Orphanet J Rare Dis 2009; 4: 9.
Swinkels ME, Simmons A, Smeets DF et al: Clinical and cytogenetic characterization of 13 Dutch patients with deletion 9p syndrome: delineation of the critical region for a consensus phenotype. Am J Med Genet A 2008; 146A: 1430–1438.
Hoischen A, van Bon BW, Rodriguez-Santiago B et al: De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet 2011; 43: 729–731.
Sobreira N, Schiettecatte F, Valle D, Hamosh A : GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 2015; 36: 928–930.
Firth HV, Richards SM, Bevan AP et al: DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 2009; 84: 524–533.
Firth HV, Wright CF,, D.D.D. Study: The Deciphering Developmental Disorders (DDD) study. Dev Med Child Neurol 2011; 53: 702–703.
Ramocki MB, Dowling J, Grinberg I et al: Reciprocal fusion transcripts of two novel Zn-finger genes in a female with absence of the corpus callosum, ocular colobomas and a balanced translocation between chromosomes 2p24 and 9q32. Eur J Hum Genet 2003; 11: 527–534.
Talisetti A, Forrester SR, Gregory D, Johnson L, Schneider MC, Kimonis VE : Temtamy-like syndrome associated with translocation of 2p24 and 9q32. Clin Dysmorphol 2003; 12: 175–177.
Shashi V, Pena LD, Kim K et al: De novo truncating variants in ASXL2 are associated with a unique and recognizable clinical phenotype. Am J Hum Genet 2016; 99: 991–999.
Kruszka P, Addissie YA, Yarnell CM et al: Muenke syndrome: an international multicenter natural history study. Am J Med Genet A 2016; 170A: 918–929.
Ettinger N, Williams M, Phillips JA 3rd : Variable expressivity and clinical heterogeneity can complicate the diagnosis and management of Pfeiffer syndrome. J Craniofac Surg 2013; 24: 1829–1832.
Solomon BD, Bear KA, Wyllie A et al: Genotypic and phenotypic analysis of 396 individuals with mutations in Sonic Hedgehog. J Med Genet 2012; 49: 473–479.
Franca MM, Orge AA, Carvalho LR et al: Novel heterozygous nonsense GLI2 mutations in patients with hypopituitarism and ectopic posterior pituitary lobe without holoprosencephaly. J Clin Endocrinol Metab 2010; 95: E384–E391.
Balk K, Biesecker LG : The clinical atlas of Greig cephalopolysyndactyly syndrome. Am J Med Genet A 2008; 146A: 548–557.
De Rubeis S, He X, Goldberg AP et al: Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014; 515: 209–215.
Iossifov I, O’Roak BJ, Sanders SJ et al: The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014; 515: 216–221.
Krumm N, Turner TN, Baker C et al: Excess of rare, inherited truncating mutations in autism. Nat Genet 2015; 47: 582–588.
Abrahams BS, Arking DE, Campbell DB et al: SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism 2013; 4: 36.
Lek M, Karczewski KJ, Minikel EV et al: Analysis of protein-coding genetic variation in 60706 humans. Nature 2016; 536: 285–291.
Samocha KE, Robinson EB, Sanders SJ et al: A framework for the interpretation of de novo mutation in human disease. Nat Genet 2014; 46: 944–950.
Acknowledgements
We are grateful to the families for consenting to participate in this publication. We thank Dr Bård Nedregaard for his input on neuroimaging studies. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003; see Nature 25533962 or www.ddduk.org/access.html for full acknowledgment). This work was supported by the intramural program of the National Human Genome Research Institute, NIH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Weiss, K., Wigby, K., Fannemel, M. et al. Haploinsufficiency of ZNF462 is associated with craniofacial anomalies, corpus callosum dysgenesis, ptosis, and developmental delay. Eur J Hum Genet 25, 946–951 (2017). https://doi.org/10.1038/ejhg.2017.86
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.86
This article is cited by
-
ZFP462 safeguards neural lineage specification by targeting G9A/GLP-mediated heterochromatin to silence enhancers
Nature Cell Biology (2023)
-
A Meta-Analysis of the Genome-Wide Association Studies on Two Genetically Correlated Phenotypes Suggests Four New Risk Loci for Headaches
Phenomics (2023)
-
Diabetes in a Weiss-Kruszka syndrome boy and a de novo deletion in 9q31.2
Endocrine (2023)
-
Genome-wide association study of cerebellar volume provides insights into heritable mechanisms underlying brain development and mental health
Communications Biology (2022)

{kind=link}
{kind=link}
{kind=link}