
Abstract
PAN2 encodes a subunit of a deadenylation complex with important functions in mRNA stability and post-transcriptional regulation of gene expression. A homozygous frameshift deletion in PAN2 was reported in a single affected individual with developmental delay and multiple congenital anomalies. Here, we describe five additional individuals from three unrelated families with homozygous predicted loss-of-function variants in PAN2. The affected individuals presented with significant overlap in their clinical features, including mild-moderate intellectual disability, hypotonia, sensorineural hearing loss, EEG abnormalities, congenital heart defects (tetralogy of Fallot, septal defects, dilated aortic root), urinary tract malformations, ophthalmological anomalies, short stature with other skeletal anomalies, and craniofacial features including flat occiput, ptosis, long philtrum, and short neck. Our data confirm that biallelic predicted loss-of-function variants in PAN2 cause a syndrome with multiple congenital anomalies, and suggest an important role of mRNA polyA tail length for proper organ formation.
Similar content being viewed by others


Introduction
Developmental disorders are of heterogeneous genetic etiology, and many of the genetic causes still remain to be unraveled [1]. Identification of several unrelated individuals harboring deleterious variants in the same gene provides evidence for disease causality and allows the recognition of clinical patterns and syndromes. Such human “knock-outs” and delineation of their phenotypic manifestations may also help unravel the basic mechanisms of human developmental biology.
Recent next-generation sequencing-based studies have established pathogenic variants affecting members of diverse RNA metabolism and processing pathways as a major cause of developmental disorders, including those involved in mRNA decay and translation initiation [2]. The majority of mammalian mature mRNAs have a long chain of adenine nucleotides (poly A tails) that are added during the RNA processing. The Pan2–Pan3 deadenylation complex shortens mRNA 3′ polyA tails, thereby regulating mRNA stability and translation efficiency. Pan2 functions as the catalytic subunit of the complex, whereas interaction with Pan3 is required for efficient mRNA binding [3]. The majority of deadenylation in the cytoplasm is carried out by one of two complexes, Pan2–Pan3 or Ccr4–Not. Despite some functional redundancy, Pan2–Pan3 was found to be more efficient at initiating deadenylation, followed by Ccr4–Not-mediated sequential shortening of the polyA tail [4,5]. Accordingly, in yeast extracts lacking Pan2p and Pan3p, transcripts were polyadenylated to lengths exceeding 90–200 adenosines [4]. In line with its important cellular function, a homozygous predicted loss-of-function variant in PAN2 was recently described in an individual affected by a complex developmental disorder with skeletal dysplasia [6]. However, the link between biallelic pathogenic PAN2 variants and developmental abnormalities is still not firmly established and the corresponding clinical picture remains largely unclear.
Here, we describe five individuals from three unrelated families with homozygous PAN2 variants, and review the previously reported case. The largely overlapping clinical features support causality and suggest that developing brain, heart, and urogenital structures are particularly vulnerable to PAN2 deficiency.
Methods
Study participants
We used GeneMatcher [7] to assemble a cohort of three unrelated families with homozygous PAN2 variants. Medical histories, pedigrees, and results of extended examinations and analyses were obtained in clinical and/or research settings in Canada, Germany, or Austria. Written informed consents for study participation and the publication of photos were obtained from all study participants or their respective guardians. The studies were approved by local ethics committees. Individual 1.1 was recruited as part of a cohort with congenital heart disease (TOF145 in [8,9,10]).
Genome-wide sequencing
Genome sequencing on whole blood from individual 1.1 was performed at The Centre for Applied Genomics (TCAG) in Toronto, Canada. Fragmented DNA (average 350 bp) was end-repaired, A-tailed and ligated with TruSeq Illumina adapters prior to library amplification. Validated libraries were pooled in equimolar quantities and sequenced on a HiSeq X platform (Illumina, CA, USA) following the manufacturer’s protocol to generate paired-end reads of 150 bases in length. Reads were mapped using the Burrows-Wheeler Aligner (BWA; GRCh37/hg19 reference sequence). The mean read depth was 33.75× (97.29% coverage at 10×, 89.46% coverage at 25×). Single-nucleotide variants and small insertions/deletions were called using GATK. Copy number variants (CNVs) were called using a modified read depth method with the programs Estimation by Read Depth with Single-nucleotide variants and CNVnator using a window size of 500 bp [11]. Variant calls were annotated using a custom pipeline developed at TCAG based on ANNOVAR. We prioritized rare, predicted protein-altering variants (minor allele frequency (MAF) < 0.1% in gnomAD v2.1.1).
For family 2, the two affected siblings (2.1 and 2.2) and their healthy parents underwent exome sequencing at the sequencing core of Helmholtz Center Munich (Munich, Germany). Paired-end 100-bp libraries were prepared from lymphocyte-derived genomic DNA and captured using the SureSelect Human All Exon v6 kit (Agilent Technologies, CA, USA) according to the manufacturer’s protocol. Samples were run on a HiSeq4000 (Illumina, CA, USA) platform, generating on average 10 Gb of uniquely mapped reads. Data processing was done with an in-house pipeline, which implements BWA for sequence mapping (GRCh37/hg19) and SAMtools, GATK, ExomeDepth, and custom scripts for variant calling and annotation. Average coverage was >100× in all samples, with 96–97% of the target sequences covered at least 20×. After quality control and elimination of sequencing artifacts, we filtered for non-synonymous variants and insertions/deletions shared by both affected subjects. The MAF threshold was set to 0.1%, using control data from gnomAD v2.1.1 and in-house exome collections (N = 20,000). As two children of consanguineous parents were affected, priority was given to homozygous alleles.
For individual 3.1, exome sequencing was performed on DNA isolated from whole blood, at the Cologne Center for Genomics, as previously described [12]. Briefly, exome sequencing was performed on two lanes of an Illumina GAIIx Sequencer using a single read 150 bp protocol after enrichment of exonic and splice-site sequences with the NimbleGen SeqCap EZ Human Exome Library v3.0. Approximately 94% of target sequences were covered at least 10× with a mean coverage of about 80×. Data analysis and processing were performed as previously described [13]. The criteria for a variant to be taken into account were: >6 reads, phred scaled quality score >15, and MAF < 0.1% according to gnomAD v2.1.1, for putative autosomal recessive variants.
Details on variant filtering are described as Supplementary material. Prioritized variants were visually assessed with the Integrative Genomics Viewer [14]. PCR and Sanger sequencing were performed for validation and co-segregation analyses. Primer pairs are available upon request. Additional methodological information is provided as Supplementary material. All variant coordinates refer to the hg19/GRCh37 human reference genome.
Results
We describe five affected individuals from three unrelated families with homozygous ultra-rare, predicted loss-of-function variants in PAN2. Notably, homozygous loss-of-function variants are absent in the gnomAD v2.1.1 dataset. Medical histories, diagnostic findings, and pedigrees are summarized in Table 1 and Fig. 1. Additional genomic variants of uncertain significance, and regions of homozygosity, are outlined in Tables S1, S2, respectively.

A Family 1. B Family 2. C Family 3. Numbers inside symbols indicate the numbers of siblings with a given phenotype. The pedigree nomenclature is in accordance with Bennett et al. [20].
Family 1
Individual 1.1 (TOF 145 in [8]), female, was born to a healthy 30-year-old mother. Pregnancy and labor were normal, and birth weight within normal limits for a term birth (3118 g, −0.62 SD). She was diagnosed with multiple congenital anomalies: tetralogy of Fallot (TOF) with atrial septal defect, hypoplastic low-lying left kidney with fetal lobulation and small left ureter, hypertrophic right kidney, and sensorineural deafness. She had mild to moderate intellectual disability (walked at 14 months and spoke 2–3 word sentences at 3 years), and attended a special education school.
Growth was initially in the normal range (4y10m: 105 cm, −0.32 SD), but became progressively delayed later in childhood (7y4m: 112.5 cm, −2.08 SD; 10y2m: 124 cm, −2.27 SD; 12y2m: 130.5 cm, −2.91 SD). Her bone age was found to be delayed at 16 years (<−2 SD below chronological age, height not documented). Her head circumference was normal (16y2m: 54.5 cm, +0.18 SD). Minor morphological features in childhood included wide-set eyes, down-slanting palpebral fissures, a high-arched palate, low hair line, short neck, broad thumbs, 2nd toes overlapping 3rd toes bilaterally, 5th toes overlapping 4th toes bilaterally.
At five years, she was admitted to the hospital because of autoimmune hemolytic anemia and idiopathic thrombocytopenic purpura and petechiae on legs and arms. A bone marrow biopsy revealed megaloblastic changes. Despite iron substitution, she remained anemic. An electroencephalogram (EEG) showed abnormal brain activity, but no spikes or wave activity characteristic of epilepsy. In adulthood, she had recurrent urinary tract infections, kidney stones, renal insufficiency, a prominent (likely enlarged) spleen, cholecystitis and cholelithiasis, pancreatitis, frequent respiratory and ear infections, a brain abscess, atrial fibrillation, and residual pulmonary stenosis after TOF repair. At age 45 years, she died of excessive bleeding after a complicated heart transplantation.
Her brother (individual 1.2) had borderline intellectual functioning, slurred speech, and temper outbursts. He had completed secondary school education but received a disability pension. He had moderate sensorineural hearing loss requiring hearing aids. In adulthood, he was diagnosed with intestinal cancer and an inguinal hernia. The facial features resembled those of his affected sister; he had down-slanting palpebral fissures, a long tubular nose, and a small face. Two sisters were healthy; two other siblings had died late in pregnancy or shortly after birth, respectively (no DNA was available) (Fig. 1).
Genome-sequencing performed on individual 1.1 revealed a homozygous stopgain variant p.(Glu779*) in PAN2 (chr12:56717114C>A; Fig. 2), which was confirmed by Sanger sequencing. The variant was also found homozygous in her affected brother, while both parents and two healthy sisters were heterozygous carriers (Fig. S1). Parents were non-consanguineous, but originated from the same region in southern Italy, consistent with two runs of homozygosity of 2.2 Mb (chr12:54778823–56984606) and 2.1 Mb (chr5:98543170–100601412) (Table S2). Since defects in mRNA deadenylation had previously been associated with telomeric shortening [15], we assessed telomere length from genome data. We did not find evidence for telomeric shortening in blood from individual 1.1 (Supplementary methods and Fig. S2).

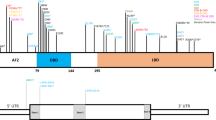
A PAN2 protein domains and variant positions, from this study (black font) and Maddirevula et al. [6] (gray font, dashed line). B Family 3 variant c.574–2A>G is predicted to result in in-frame skipping of exon 5, encoding a conserved repeat within the WD40 domain (amino acid residues 192 to 217).
Family 2
Individual 2.1, a 20-year-old young woman, is the first child of healthy consanguineous parents of Turkish descent. Pregnancy and delivery at term were normal, as were birth measurements (weight 3000 g, −0.88 SD; length 50 cm, −0.19 SD). After birth, a cleft palate was noted but treated only at age 6 years. A congenital heart defect (TOF) was identified which required surgical repair. In addition, she had an operation of an anovesical fistula in her first year of life. She had feeding difficulties related to muscular hypotonia. Between age 6 months and 3 years, psychomotor developmental delay became evident. She walked at 4 years of age and spoke only a few words. Ophthalmologic investigations at age 6 showed hyperopic astigmatism left more than right and strabismus convergens left. EEG revealed epileptiform discharges but she never demonstrated clinical seizure activity. Brain MRI at age 7 years was normal. At age 10–15 years, moderate sensorineural hearing loss was diagnosed. By age 17 years, she had acquired meaningful speech, and she attended a school for individuals with intellectual disability. From age 19 she worked as a kitchen and hairdresser’s assistant and was well integrated into her family and workplace. Neurologic examination noted abnormal fine motor function and gait imbalance. A number of morphological features such as short neck, broad nasal root, hypertelorism, low set ears, bowed and prominent eyebrows, synophrys, short terminal phalanges with hypoplastic nails, and clinodactyly of the fifth fingers were observed (Fig. 3A–E, O, S). When last documented at age 16 years, her height was markedly reduced (141.1 cm, −4 SD), along with weight (34.7 kg, −4.3 SD), while her head circumference was normal (53.5 cm, −0.75 SD).

Individual 2.1 at age 6 years (A, B, O), 16 years (C, D, S), and 20 years (E). Individual 2.2 at age 3 years (F, G), 13 years (H, I, P), and 17 years (J). Individual 3.1 at age 1 year 9 months (K, Q, R), 2 years 10 months (L, M), and 8 years 10 months (N).
Her brother (individual 2.2), a 17-year-old boy born without perinatal problems and with normal birth measurements (weight 3800 g, +0.49 SD; length 50 cm, −0.52 SD), showed a constellation of symptoms similar to those of his older sister. He had a bifid uvula but no overt clefting. Cardiologic examinations revealed a mild dilatation of the aortic root (30 mm, +2.4 SD). When last documented at 13.5 years, he had normal growth parameters (height: 149 cm, −1.37 SD; weight: 51 kg, +0.28 SD; head circumference: 55.5 cm, +0.71 SD). He had sensorineural hearing loss, intellectual disability, and morphological features similar to his sister (Fig. 3F–J,P). Additionally, he developed myoclonic epilepsy in early infancy which was controlled with valproate. At age 13 years, his brain MRI was normal. He was diagnosed with strabismus alternans and hyperopia (+5/+7 diopters). He made slow progress in motor and language functions and had acquired simple speech at age 16 years.
The siblings were diagnosed with a syndromic neurodevelopmental disorder; however, no specific abnormalities were detected on exhaustive biochemical and molecular standard testing. Ultimately, exome sequencing of the entire family quartet was undertaken in a research context. The analysis prioritized a homozygous nonsense variant, c.3408dupT, p.(Glu1137*) in PAN2 (Fig. 2), shared by both affected siblings. One sister was unaffected; another sister had died early in infancy (no DNA was available) (Fig. 1).
Family 3
Individual 3.1, an 8-year-old girl, is the first-born child of healthy consanguineous parents. A younger sister was unaffected. Labor was complicated by shoulder dystocia and perinatal asphyxia; she was born at term with normal weight (3390 g, −0.05 SD), length below average (47 cm, −1.51 SD), and small head circumference (32 cm, −1.9 SD). Soon after birth, right renal agenesis and left double kidneys were diagnosed on ultrasound. Her early postnatal period was complicated by persistent granulocytopenia, neutropenia, and hyporegenerative anemia. Recurrent hypoglycemia, lactic acidosis, and hepatomegaly were suggestive of a glycogen storage disease type I (GSD1B). This diagnosis was confirmed by identification of a homozygous, known pathogenic variant in SLC37A4, c.1015G>T, p.(Gly339Cys).
In addition, the girl had several clinical signs and symptoms that were likely to be unrelated to GSD1B, including failure to thrive despite adequate diet, progressive growth delay, microcephaly, muscular hypotonia, global developmental delay, sensorineural hearing loss, strabismus, ptosis, Rieger anomaly and hyperopia, ectodermal features with recurrent neurodermatitis, sparse hair and delayed teeth eruption, and hyperparathyroidism. She first developed febrile seizures upon receiving a pneumococcal vaccine at the age of 16 months, followed by some additional febrile seizures until the age of 2 years.
The patient presented at the clinical genetics department of the University Medical Center Hamburg-Eppendorf at the age of 22 months. She was a friendly girl with no speech and no independent walking, with distinct craniofacial features including a flat occiput, sparse hair and eyebrows, broad forehead, deep-set and large eyes, long philtrum, thin upper lip, and low-set and small, dysmorphic ears. Her hands and feet were small with loose skin and clinodactyly of the fifth fingers. Due to feeding difficulties, she was fed by nasogastric tube. Her growth was poor with a height of 78 cm (−2.32 SD), weight of 10 kg (−1.44 SD), and OFC of 44.5 cm (−3.81 SD). She acquired independent walking after the second year of life and spoke first words at 3 years. Her further disease course was complicated by GSD1B-associated symptoms, including recurrent hypoglycemia (likely due to feeding difficulties) and neutropenia, and poor response to a combined corticosteroid/granulocyte colony-stimulating factor therapy. Consequently, and due to splenomegaly with recurrent severe thrombocytopenia, a partial spleen embolization had been performed with a transient effect. The complicated course of GSD1B eventually lead to liver transplantation at the age of three years. After a good initial recovery, the transplanted liver started to fail at the age of 6 years, leading to a severe liver failure and a second liver transplantation at the age of 8 years and 2 months. At the age of 8 years and 4 months, she was recovering well. Eating capabilities improved albeit she ate mostly pureed food. She acquired new gross and fine motor skills and showed improved concentration and broader interests. Her height was 105 cm (−4.68 SD), weight was 15 kg (−5.72 SD) and OFC was 47.5 cm (−3.94 SD). No signs of skeletal dysplasia were found on X-rays at the age of 8 years. She still had severe hyperopia, glaucoma and Rieger anomaly, severe to profound bilateral hearing impairment, and ectodermal features including sparse hair, dry skin, gingiva hyperplasia and teeth anomalies. Craniofacial features included hypertelorism, slightly down-slanting palpebral fissures, deep-set eyes, poor modulated philtrum, small upper vermillion, dysplastic teeth, microgenia, and low-set dysmorphic ears (Fig. 3K–N, Q, R). She spoke only simple sentences and had several autistic features, like repetitive movements when excited or upset, and compulsive behavior.
Exome sequencing was performed on the proband. This analysis and subsequent Sanger sequencing-based segregation analyses with DNA samples of the proband, both parents and her unaffected sister, revealed rare biallelic variants in six genes (Table S1), in addition to the previously identified SLC37A4 pathogenic variant. Of these, only the c.574–2A>G variant in PAN2 was predicted to have severe impact on protein structure (Fig. 2, Tables S1, S3).
Discussion
We describe five affected individuals from three unrelated families harboring previously unreported rare homozygous predicted loss-of-function variants in PAN2. The associated phenotypes, and that of the single case reported by Maddirevula et al. [6], were largely overlapping. Collectively, they indicate a clinical syndrome, characterized by developmental delay/intellectual disability in all affected individuals, sensorineural hearing loss in five of six individuals, and incompletely penetrant congenital anomalies: heart defects in four of six individuals (TOF, septal defects, aortic root dilation), urinary tract malformations in four of six individuals (kidney hypoplasia or agenesis, anovesical fistula), and congenital ophthalmological anomalies in two of six individuals (Rieger anomaly, posterior embryotoxon, maculopathy). Four of six individuals had a history of seizures or EEG anomalies, not necessarily requiring anticonvulsant treatment. Craniofacial features included high or cleft palate/bifid uvula, flat occiput, ptosis, hypertelorism, tubular nose/broad nasal root, long philtrum, low hair line, low-set ears, and short neck in at least two unrelated probands. Short stature along with other skeletal anomalies (metopic craniosynostosis, scoliosis, delayed bone age, broad thumbs, clinodactyly) were described in multiple individuals, however comprehensive physical or radiographic evaluation for skeletal dysplasia was not performed in this study group. Individual 3.1 had a dual diagnosis of PAN2-related disease and GSD1B, which resulted in what is likely a blended phenotype, particularly regarding growth, gastrointestinal, renal, and hematological anomalies.
Three of the total four rare homozygous PAN2 variants (Table 1) were predicted to result in a premature stop codon and nonsense-mediated mRNA decay (predicted loss-of-function variants; Fig. 2A). The fourth variant, c.574–2A>G (Family 3), affected a canonical splice acceptor site, and several splice prediction tools (NetGene2, MutationTaster and Splice Prediction by Neural Network) predicted this variant to abolish the splice acceptor site. Thus, variant c.574–2A>G likely results in in-frame skipping of exon 5, encoding a conserved repeat within the WD40 domain (Fig. 2B). In yeast, the WD40 domain was reported to sense the length of the polyA tail, and recombinant Pan2 lacking the WD40 domain resulted in severely impaired ability to deadenylate mRNA 90A tails, similar to strains with a complete Pan2 deletion [16]. PAN2 loss-of-function is therefore the most likely disease mechanism in the reported families (supporting evidence for pathogenicity is summarized in Table S3). Notably, no homozygous loss-of-function variant in PAN2 was deposited in the gnomAD dataset. Moreover, PAN2 has a high probability of being loss-of-function intolerant (gnomAD pLI score of 0.94, and a low observed / expected (oe) metric of 0.19 (0.12–0.32)).
By regulating translation and initiating mRNA decay, deadenylation of mRNA is an important mechanism in the post-transcriptional regulation of gene expression [17]. Perturbations of mRNA metabolism and decay are well-established causes of neurodevelopmental disorders and intellectual disability [2]. In general, mRNA deadenylation requires sequential involvement of Pan2/Pan3 (for the initiation of deadenylation) and the Ccr4-Not complex (for further shortening of polyA tails). Partial functional redundancy between the two complexes may mitigate the consequences of PAN2 loss-of-function in humans. This conclusion is supported by studies of Pan2Δ, Ccr4Δ, and Pan2Δ/Ccr4Δ yeast strains, where the most severe phenotypes and a complete inability to shorten mRNA polyA tails were observed in the double mutant [18]. Pan2 mouse mutants had increased heart weight and increased eosinophil cell numbers (mice homozygous for a mutant allele induced through N-ethyl-N-nitrosourea were embryonically lethal; http://www.informatics.jax.org/marker/MGI:1918984).
In contrast to the recessively inherited phenotype of PAN2 deficiency described here, alterations affecting subunits of the CCR4–NOT complex cause rare autosomal dominant developmental disorders: (i) syndromic holoprosencephaly/Vissers-Bodmer syndrome (CNOT1; OMIM-P #619033), (ii) neurodevelopmental disorder with nasal speech, dysmorphic facies, and variable skeletal anomalies (CNOT2; OMIM-P #618608), and (iii) neurodevelopmental disorder with speech delay, autism, and dysmorphic facies (CNOT3; OMIM-P #618672). Although the pathogenic mechanisms have yet to be unraveled for those diseases, impaired mRNA deadenylation may contribute to the phenotype. Defects in mRNA deadenylation were found in individuals with dyskeratosis congenita (PARN; OMIM-P #616353), a rare autosomal recessive disorder characterized by short telomeres, skin abnormalities, bone marrow failure, and a high risk of developing cancer. Due to the link between mRNA deadenylation and telomere disease [15,19] and since affected individuals from two families (1.1, 1.2, 3.1) were reported to have thrombocytopenia and anemia, we studied telomere lengths in the genome sequencing data of individual 1.1. However, we did not find evidence for telomeric shortening (Fig. S2).
Limitations
Three of the four families now reported with biallelic PAN2 variants, including the previous report [6], were consanguineous (Table 1). Offspring of consanguineous parents typically have numerous rare, predicted damaging homozygous variants. Except for family 3 with GSD1B, we did not identify other pathogenic or likely pathogenic variants (a list of variants of uncertain significance is provided in Table S1). However, regions of homozygosity of variable length and extent, including the region overlapping PAN2, were present in all four families (Table S2). For families 1 and 3, de novo variants could not be excluded, as sequencing was performed for the probands only. Other unassessed genetic background may also affect variable expression of features associated with rare homozygous PAN2 variants. Premature (early infant) mortality was a prominent feature in two families (Fig. 1) but molecular data would be needed to confidently ascribe this to PAN2 variants. As for the previously published individual [6], we did not functionally assess deadenylation in cells from affected individuals.
Conclusions
Homozygous PAN2 predicted loss-of-function variants are associated with an autosomal recessive syndrome (PAN2-related multiple congenital anomalies syndrome), which is characterized by intellectual disability, sensorineural hearing loss, short stature, variable craniofacial features, and incompletely penetrant congenital anomalies of the heart, genitourinary tract, and eyes. While our data support an important function of PAN2 in human development, providing further link between syndromic intellectual disability and regulation of mRNA translation [2], the underlying molecular mechanisms and the full clinical spectrum remain to be characterized further.
Data availability
Data generated and analyzed during this study can be found within the published article and its supplementary files, and in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/; Accession VCV001342924, VCV001342923, VCV001342925). Additional data are available from the corresponding author on reasonable request.
References
Kaplanis J, Samocha KE, Wiel L, Zhang Z, Arvai KJ, Eberhardt RY, et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586:757–62.
Weil D, Piton A, Lessel D, Standart N. Mutations in genes encoding regulators of mRNA decapping and translation initiation: links to intellectual disability. Biochem Soc Trans. 2020;48:1199–211.
Wolf J, Valkov E, Allen MD, Meineke B, Gordiyenko Y, McLaughlin SH, et al. Structural basis for Pan3 binding to Pan2 and its function in mRNA recruitment and deadenylation. EMBO J. 2014;33:1514–26.
Brown CE, Sachs AB. Poly(A) tail length control in Saccharomyces cerevisiae occurs by message-specific deadenylation. Mol Cell Biol. 1998;18:6548–59.
Yamashita A, Chang TC, Yamashita Y, Zhu W, Zhong Z, Chen CY, et al. Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat Struct Mol Biol. 2005;12:1054–63.
Maddirevula S, Alsahli S, Alhabeeb L, Patel N, Alzahrani F, Shamseldin HE, et al. Expanding the phenome and variome of skeletal dysplasia. Genet Med. 2018;20:1609–16.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–30.
Silversides CK, Lionel AC, Costain G, Merico D, Migita O, Liu B, et al. Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet. 2012;8:e1002843.
Reuter MS, Jobling R, Chaturvedi RR, Manshaei R, Costain G, Heung T, et al. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet Med. 2019;21:1001–7.
Manshaei R, Merico D, Reuter MS, Engchuan W, Mojarad BA, Chaturvedi R, et al. Genes and pathways implicated in tetralogy of fallot revealed by ultra-rare variant burden analysis in 231 genome sequences. Front Genet. 2020;11:957.
Trost B, Walker S, Wang Z, Thiruvahindrapuram B, MacDonald JR, Sung WWL, et al. A Comprehensive workflow for read depth-based identification of copy-number variation from whole-genome sequence data. Am J Hum Genet. 2018;102:142–55.
Lessel D, Ozel AB, Campbell SE, Saadi A, Arlt MF, McSweeney KM, et al. Analyses of LMNA-negative juvenile progeroid cases confirms biallelic POLR3A mutations in Wiedemann-Rautenstrauch-like syndrome and expands the phenotypic spectrum of PYCR1 mutations. Hum Genet. 2018;137:921–39.
Lessel D, Wu D, Trujillo C, Ramezani T, Lessel I, Alwasiyah MK, et al. Dysfunction of the MDM2/p53 axis is linked to premature aging. J Clin Investig. 2017;127:3598–608.
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6.
Tummala H, Walne A, Collopy L, Cardoso S, de la Fuente J, Lawson S, et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Investig. 2015;125:2151–60.
Schafer IB, Yamashita M, Schuller JM, Schussler S, Reichelt P, Strauss M, et al. Molecular basis for poly(A) RNP architecture and recognition by the Pan2-Pan3 deadenylase. Cell. 2019;177:1619–31 e21.
Wiederhold K, Passmore LA. Cytoplasmic deadenylation: regulation of mRNA fate. Biochem Soc Trans. 2010;38:1531–6.
Tucker M, Valencia-Sanchez MA, Staples RR, Chen J, Denis CL, Parker R. The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell. 2001;104:377–86.
Mason PJ, Bessler M. mRNA deadenylation and telomere disease. J Clin Investig. 2015;125:1796–8.
Bennett RL, French KS, Resta RG, Doyle DL. Standardized human pedigree nomenclature: update and assessment of the recommendations of the National Society of Genetic Counselors. J Genet Couns. 2008;17:424–33.
Acknowledgements
We thank the patients and their families for participation in this study.
Funding
This work was funded in part by a donation from the W. Garfield Weston Foundation (ASB), and a grant by the Deutsche Forschungsgemeinschaft (LE 4223/3–1 to D.L.). SWS is funded by the Glaxo Smith Kline-CIHR Chair in Genome Sciences at the University of Toronto and The Hospital for Sick Children. ASB holds the Dalglish Chair in 22q11.2 Deletion Syndrome at the University Health Network and University of Toronto.
Author information
Authors and Affiliations
Contributions
MSR, MZ, MH, JA, HT, BT, CK, SWS, ASB, and DL were involved in the generation, analysis and interpretation of the genomic data. MSR, MH, TH, LP, RS, CK, SR, ASB, and DL reviewed the clinical data. MSR, MZ, MH, ASB, and DL wrote the manuscript. All authors revised the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
SWS serves on the Scientific Advisory Committees of Population Diagnostics and Deep Genomics, and is a Highly Cited Academic Advisor to the King Abdulaziz University. The other authors declare no conflicts of interest.
Ethical approval
Informed consents were obtained from all probands and/or their legal guardians. The study was conducted under the Declaration of Helsinki principles and approved by the local Research Ethics Boards (Research Ethics Boards at the Toronto University Health Network (REB 98-E156), Centre for Addiction and Mental Health (REB 154/2002), Ethics Committee of the Hamburg Chamber of Physicians: PV 3802, Medical University of Innsbruck, and Technical University of Munich).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Reuter, M.S., Zech, M., Hempel, M. et al. Biallelic PAN2 variants in individuals with a syndromic neurodevelopmental disorder and multiple congenital anomalies. Eur J Hum Genet 30, 611–618 (2022). https://doi.org/10.1038/s41431-022-01077-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01077-y
This article is cited by
-
No gene to predict the future?
European Journal of Human Genetics (2022)
