
Abstract
Congenital cataract (CC) is one of the most important causes for blindness or visual impairment in infancy. A substantial proportion of isolated CCs has monogenic causes. The disease is genetically heterogeneous, and all Mendelian modes of inheritance have been reported. We mapped a locus for isolated CC on 19p13.1–q13.2 in a distantly consanguineous German family with two sisters affected by dense white cataracts. Whole-exome sequencing identified a homozygous nonsense variant c.4489C>T (p.(R1497*)) in SIPA1L3 (signal-induced proliferation-associated 1 like 3) in both affected children. SIPA1L3 encodes a GTPase-activating protein (GAP), which interacts with small GTPases of the Rap family via its Rap-GAP-domain. The suggested role of Rap GTPases in cell growth, differentiation and organization of the cytoskeleton in the human lens, and lens-enriched expression of the murine ortholog gene Sipa1l3 in embryonic mice indicates that this gene is crucial for early lens development. Our results provide evidence that sequence variants in human SIPA1L3 cause autosomal recessive isolated CC and give new insight into the molecular pathogenesis underlying human cataracts.
Similar content being viewed by others

Introduction
Congenital cataract (CC) is the most common ocular disease leading to blindness or severe visual impairment in children worldwide. The human lens is derived from epithelial cells, which differentiate into fiber cells to form the bulk of the lens. A single layer of epithelial cells covers the anterior surface of the lens and regulates most of its homeostatic functions. Any disturbance of lens development or homeostasis can result in cataract. CC may be isolated, associated with other anterior segment malformations of the eye, or other systemic findings as part of a syndrome.1 In developed countries about one-third of isolated CCs are hereditary, and all three forms of Mendelian inheritance have been observed.2 More than 20 different genes have been identified that cause either isolated congenital or isolated juvenile cataract (Table 1). They can be divided into four major groups: genes encoding (a) crystallins, (b) membrane proteins, (c) cytoskeletal proteins, and (d) transcription factors or other proteins regulating gene expression. Sequence variants in genes coding for crystallins (eg, CRYAA), the main structural proteins of the lens, account for ~50% of isolated familial cataracts.3, 4, 5, 6, 7, 8, 9, 10, 11, 12 Genes of group (b) encode gap junction proteins (GJA3, GJA8), other cell junction proteins (LIM2, TMEM114), aquaporin water channels (MIP), and proteins involved in membrane-associated transport or signaling (eg, CHMP4B, EPHA2).13, 14, 15, 16, 17, 18 Genes of group (c) are essential for the synthesis of intermediate filament, and intermediate filament-like proteins (VIM, BFSP1, BFSP2).19, 20, 21 Sequence variants in genes of group (d) are often associated with a syndromic phenotype and other ocular defects (eg, PITX3).22, 23, 24 The remaining genes cannot be classified in the four major groups. They often encode proteins with unexpected functions, for example, those involved in lipid metabolism (AGK).25 However, even more genes are expected to be involved in the etiology of CC. Their discovery could provide new insight into the underlying pathomechanisms.
We studied a consanguineous family with two sisters affected by isolated CC, indicating autosomal recessive inheritance by linkage analysis and whole-exome sequencing (WES).
Materials and methods
Patient ascertainment and DNA isolation
The two patients, their healthy sister and parents were seen at the Department of Ophthalmology of St Vincentius-Kliniken, Karlsruhe, Germany, and the Institute of Human Genetics, Heidelberg, Germany. Parents gave written consent for publication. The study adhered to the tenets of the Declaration of Helsinki and was approved by the local ethics committee. Genomic DNA of patients, their sister and parents was isolated from blood as previously described.26
Array analysis/SNP genotyping
Affymetrix CytoScan HD Oligo/SNP-Array was performed according to the manufacturer's instructions to genotype DNA of the affected individuals and both parents, and to exclude genomic imbalances in the affected patients. Arrays were scanned with the AffymetrixGeneChip Scanner 3000 7G and analyzed with Affymetrix Chromosome Analysis Suite software version 1.2 and Annotation Net Affx Build 32. Analysis for genomic imbalances was done at a resolution of 100 kb. Interpretation was based on human reference sequence GRCh37/hg19, February 2009.
Statistical linkage analysis
Genome-wide parametric linkage analysis with SNP genotypes was performed using ALOHOMORA and MERLIN software,27, 28 assuming affected family members were homozygous at a putative disease locus for an autosomal recessive disease allele inherited from a common ancestor. After performing several data quality checks, SNP marker with minor allele frequency (MAF) 0.15 and a minimum distance of 100 000 bp were selected to ensure low linkage disequilibrium between the markers.
Whole-exome sequencing
We performed WES on the patients (I.2 and I.3) and their parents (II.1 and II.2). Exome capturing was done using Agilent SureSelect Human All Exon V4 (without UTR). The samples were sequenced to an average base coverage of 145 × on targeted regions, with 99.038% of the targeted bases having at least 10 × coverage. For each sample, the raw reads were mapped to the hs37d5 (hg19+decoy sequences) reference genome using BWA 0.6.229 and PCR duplicates were marked by Picard (http://picard.sourceforge.net/). SNVs and short indels were called using SAMtools30 and Platypus,31 respectively, and annotated using ANNOVAR.32 In-house Perl scripts were used to filter variants according to the following criteria: minimum coverage of 10 reads, a minimum QUAL score of 20 for SNVs, and ‘PASS’ for the built-in filters from Platypus for Indels. Variants with >1% MAF in the 1000 genome project33 were considered as common alleles and removed from the candidate list. An in-house database with 79 exomes whose phenotype are different from the CC, were used to filter the sequencing artifacts and common alleles. Variants were removed if they were present with the same genotype in the in-house database. Using the ANNOVAR annotations, coding variants were defined as missense or nonsense variants, indels overlapping exonic regions, and variants ±2 base around the intron–exon junction (splice sites). Variants reported in this study have been submitted to the LOVD database (http://databases.lovd.nl/shared/genes, IDs 00025020, 00025118, 00025124, 00025125).
Sanger sequencing
Validation of the SIPA1L3 sequence variant was performed by Sanger sequencing. Exon 17 and adjacent intron boundaries of the SIPA1L3 gene (RefSeq NM_015073.1, exon numbering according to Ensembl database transcript ENST00000222345 (http://www.ensembl.org)) were sequenced from affected individuals (I.2 and I.3) and both parents (II.1 and II.2) using Big Dye Terminator V1.1 cycle sequencing kit and ABI 3130xl genetic analyzer. Sanger sequencing was also performed on genomic DNA from the healthy sister (I.1), to exclude homozygosity for the SIPA1L3 nonsense variant. Primer sequences and PCR conditions are available on request.
cDNA analysis of SIPA1L3
RNA was isolated form peripheral blood of patient I.2 and healthy control persons as previously described.34 cDNA was synthesized through reverse transcription (RevertAid H Minus Reverse Transcriptase, Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer's instructions.
Different regions of the SIPA1L3 cDNA were amplified by PCR. Sequence of primers and PCR conditions are available on request. PCR products were assayed on an agarose gel.
Results
Patient reports
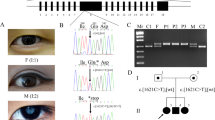
The parents were of German ancestry and consanguineous (fourth-degree cousins, II.1 and II.2, Figure 1a). Their first child was a healthy girl (I.1, Figure 1a) with normal development and without ocular problems. In the second child (I.2, Figure 1a), bilateral dense white cataracts were diagnosed at age 2 weeks. Blood and urine analyses for metabolic diseases were normal. The third child (I.3, Figure 1a) was diagnosed with bilateral dense white cataracts soon after birth. Pregnancy and birth history of all three children were unremarkable, there was no history of intrauterine infections or maternal medication. Both affected children were treated by cataract surgery, and aphakia was corrected with contact lenses. On last follow-up at age 4 years (I.2) and 2 years (I.3), both children showed a normal psychomotor development, normal growth, and no dysmorphic signs. Ophthalmologic examination of both parents did not show any ocular abnormalities.

(a) The predigree of the family: extended family history showed distant consanguinity of the parents. (b) Linkage analysis: LOD-score distribution relative to chromosomal location with the highest LOD-score region on chromosome 19 (40.2–66.0 cM, corresponding to 17.0–39.2 Mbp). The lower peak on chromosome 3 does not reach a level of genome-wide significance.
Array analysis showed no significant copy number aberrations
The affected children (I.2 and I.3) were screened for CNVs (copy number variants). A paternally inherited duplication chr6.hg19:g.57237993_57641858dup was detected in patient I.2, but not in her affected sister (I.3). In both children no other CNVs (deletions or duplications not previously listed in the Database of Genomic Variants (http://dgv.tcag.ca/)) were found across the genome.
Genotyping showed a common haplotype block on chromosome 19p13.11–q13.2
Linkage analysis demonstrated genome-wide significant evidence of linkage of disease to a locus on chromosome 19p13.11–q13.2 (ranging from 16 743 209 to 39 150 199 bp, between SNP markers rs12461484 and rs7351086). The locus showed a maximal odds ratio lod-score of 3.3 (Figure 1b). The identified haplotype block on chromosome 19p13.11–q13.2 contained 271 protein coding genes in GENCODE version 19 (http://www.gencodegenes.org/), but none of the previously identified genes for isolated autosomal recessive CC.
WES revealed a homozygous nonsense variant in SIPA1L3
Subsequent WES of the patients and their parents showed only three coding variants (in the genes CYFIP1, NPHS1, and SIPA1L3), which were homozygous in both affected siblings (I.2 and I.3) and heterozygous in their parents (II.1 and II.2), and therefore consistent with an autosomal recessive disease model in a consanguineous family. The variant in CYFIP1 (c.2707G>A; p.(G903S), RefSeq NM_014608.3) is listed in the dbSNP database (rs139635799, http://www.ncbi.nlm.nih.gov/SNP,35) and in the Exome Variant Server (EVS, http://evs.gs.washington.edu/EVS/,36) with a MAF of 0.592% and is therefore unlikely to be the cause of the cataract. This variant is not located within the linkage region on chromosome 19.
The variants affecting NPHS1 and SIPA1L3 were located in the linkage region of chromosome 19 (17.9–39.2 Mbp). The variant in NPHS1 (c.791C>G; p.(P264R), RefSeq NM_004646.3) is suggested to be a benign variant based on its allele frequency (MAF of ~1% according to the dbSNP database (rs34982899), and MAF of 1.5698% in the EVS) and on prediction by Mutation Taster (http://www.mutationtaster.org)37 and SIFT analysis (http://sift.jcvi.org/).38 In addition, sequence variants in NPHS1 affecting gene function cause the severe phenotype of autosomal recessive congenital nephrotic syndrome of the Finnish type, which can clinically be excluded in our patients.
The variant in SIPA1L3 (c.4489C>T; p.(R1497)*, RefSeq NM_015073.1) is absent in the 1000 genome project39 and in the EVS, suggesting it is extremely rare in the population. It is predicted to introduce a premature stop codon in exon 17 at amino acid 1497 (p.(R1497*)). The homozygous SIPA1L3 variant was validated by Sanger sequencing in both affected children. Both parents were confirmed to be heterozygous carriers (Figure 2a). Sanger sequencing on genomic DNA from the healthy sister (I.1) excluded homozygosity for the SIPA1L3 variant (data not shown).

(a) Sequence analysis by Sanger sequencing confirmed homozygosity for the nonsense mutation c.4489C>T (p.R1497*) in exon 17 of SIPAL3 in the two affected siblings (II.2 and II.3) and heterozygosity in their healthy parents (I.1 and I.2). (b) Exon/intron structure of SIPA1L3. Exons are shown as vertical lines. Exon numbering is according to ensemble transcript ENST00000222345 (http://www.ensembl.org). The SIPA1L3 variant c.4489C>T (marked by an arrow) is located in exon 17 of the full-length mRNA (NM_015073.1, transcript ENST00000222345), which comprises 22 exons. The first two exons are non-coding exons. The Ensemble database lists 12 putative transcripts, but only the full-length transcript (ENST00000222345) is known to be protein coding. (c) Schematic domain structure of SIPA1L3 protein and its paralogs SIPA1, SIPA1L1 and SIPA1L2. The location of the SIPA1L3 mutation c.4489C>T (p.R1497*) is shown by an arrow. Conserved protein domains of SIPA1, SIPA1L1, SIPA1L2 and SIPA1L3 are a Rap GTPase-activating protein (Rap-GAP) domain, a PDZ domain, and coiled-coil (CO) domain. The domain of unknown function (DUF3401) is similar between SIPA1L1, SIPA1L2 and SIPA1L3 but not present in SIPA1. (c and d) cDNA analysis of SIPA1L3. Amplification of exons 11–13 and 16–18 of SIPA1L3 cDNA, respectively. 1–3 and 8–10: cDNA of healthy control persons; 4 and 11: genomic DNA of healthy control person; 5 and 12: cDNA of the patient; 6 and 13: negative control; 7: marker (100 bp ladder).
The SIPA1L3 variant does not result in nonsense-mediated mRNA decay (NMD)
We were able to amplify cDNA of two different regions of SIPA1L3 in patient I.2 (exons 11–13 and 16–18, Figure 2d), showing that SIPA1L3 mRNA was not subject to degradation by NMD. This indicates that the SIPA1L3 variant results in premature termination of translation and in the synthesis of a truncated protein lacking the last 284 amino acids.
Discussion
We identified a nonsense variant (c.4489C>T; p.(R1497*)) in the gene SIPA1L3 (signal-induced proliferation-associated 1 like 3) in a consanguineous family with autosomal recessive CC using a combined strategy of linkage analysis with WES.
In the present family with parents who were 4th degree cousins, a single region with significant linkage could be identified (19p12.11–q13.2, lod-score: 3.3). This illustrates that, in particular, in case of distant consanguinity linkage analysis may help to bring down the number of genetic variants identified by high throughoutput sequencing. Within the linkage region, WES identified only two homozygous sequence variants (NPHS1: c.791C>G; p.(P264R) and SIPA1L3: c.4489C>T; p.(R1497*)) in the affected individuals. The variant in NPHS1 was excluded from further considerations because of its frequency and predicted effect. The SIPA1L3 variant c.4489C>T introduces a premature stop codon at amino acid 1497 p.(R1497*). SIPA1L3 full-length mRNA (NM_015073.1) encodes 1781 amino acids. The nonsense variant was predicted to be disease causing by in silico analysis and expected to result either in NMD or a premature termination of translation. Expression analysis in patient 1 showed that SIPA1L3 mRNA is not subject to NMD, indicating synthesis of a truncated protein lacking the last 284 amino acids.
This is the first variant in SIPA1L3 that has been described in a human disease. SIPA1L3 has been previously identified as a putative candidate gene for isolated CC in a study using mainly a mouse embryonic gene expression data set. SIPA1L3 showed highly lens-enriched expression patterns at three key developmental time points from lens placode invagination to lens primary fiber cell differentiation.40 The early expression of Sipa1l3 in murine lens and the presence of a complete white CC, which represents the end stage of cataract formation, in the present family both indicate that sequence variants in SIPA1L3 might have a very early effect on lens development, probably at the time of fiber cell differentiation.
SIPA1L3 falls within a previously mapped human cataract locus in which the causative gene had not been identified.41 Sequencing of several candidate genes within this region (MIP, LIM2, SIX5, and FTL, but not SIPA1L3) in the affected family members failed to detect a sequence variant. However, in this family the cataract showed autosomal dominant inheritance, later onset and a cortical localization.41 Other putative cataract genes in the previously mapped region for autosomal dominant cataract include PRX, EML2, SPINT2, and PVRL2.40 The above candidate genes were covered in our exome sequence data and included for further data analysis but did not yield any interesting candidate variants. For the time being, it cannot be excluded that sequence variants in SIPA1L3 may result in both autosomal dominant and autosomal recessive CC, probably with a more severe phenotype and earlier onset in homozygous individuals than in heterozygous ones.
The protein encoded by SIPA1L3 (signal induced proliferation associated protein 1 like 3, SIPA1L3, alias SPAL3) is conserved among different species (Supplementary Table S1). Human SIPA1L3 is predicted to contain a Rap GTPase-activating protein (Rap-GAP) domain, a PDZ domain, a domain of unknown function (DUF3401), and a C-terminal coiled-coil domain (Figure 2c). SIPA1L3 is closely related to SIPA1 (alias SPA1), SIPA1L1 (alias SPAL1), and SIPA1L2 (alias SPAL2) (Figure 2c, Supplementary Figure S2). The presence of a Rap-GAP domain indicates that SIPA1L3 is a GAP for small G-proteins of the Rap family. Binding of Rap to the Rap-GAP domain leads to conversion of Rap from its active, GTP-bound form to the inactive, GDP-bound form. This mechanism has been well studied for SIPA1 and SIPA1L1, which are negative regulators of Rap1 (RAP1A, Ras-related protein Rap-1A) and Rap2 (RAP2A).42, 43
Rap1 and other small GTPases are suggested to be involved in important regulatory functions in the developing lens, possibly related to epithelial cell proliferation, fiber cell differentiation, gap junction formation, and cytoskeletal organization.44 Interestingly, Rap1 controls integrin activation and integrin-mediated cell adhesion in different cell types. Integrins also regulate lens epithelial cell proliferation, survival and differentiation into fiber cells (for review see45).
The role of the PDZ domain, which is predicted to be required for protein–protein interactions, the C-terminal DUF3401 and the coiled-coil domain of SIPA1L3 is still unknown. In silico network analyses using Ingenuity software46 showed an interaction between SIPA1L3 and 14-3-3ζ (YWHAZ), a small intracellular signaling molecule identified as one of the key proteins in human lens,47 and also indicated an interaction between Rap1 and 14-3-3ζ. Gene expression data of rat lens indicated that several isoforms of 14-3-3 (including 14-3-3ζ) might be involved in cataract formation.48 Furthermore, the PDZ domain of the SIPA1L3 homolog SIPA1L1 interacts with the renal aquaporine 2 water channel.49 This might be of interest, because sequence variants in a gene for another aquaporine (aquaporine 0, AQPO, alias MIP) cause cataract formation.13 In neuronal cells, an interaction between the PDZ domain of rat SIPA1L1 (SPAR) and the EphA4 receptor has been demonstrated.50
The SIPA1L3 variant c.4489C>T; p.(R1497*) is predicted to result in a shortened protein, which includes the Rap-GAP and PDZ domain but lacks most parts of the DUF3401, the coiled-coil domain and the neighboring seven C-terminal amino acids (Figure 2). The exact function of the highly conserved C-terminal coiled-coil domain of SIPA1L3 has not been elucidated so far. Previous studies of the homolog SIPA1L1 have shown that its C-terminal part interacts with casein kinase I epsilon (CKIɛ), a protein involved in Wnt signaling. CKIɛ stimulates SIPA1L1 degradation, and alleviates SIPA1L1-mediated Rap1 inhibition.51 If SIPA1L3 also interacts with CKIɛ, a C-terminal truncated SIPA1L3 protein might escape CKIɛ-induced degradation. Otherwise, it is possible that truncation of SIPA1L3 leads to aberrant protein folding, resulting in loss-of-function. We would expect that both mechanisms result in disturbance of SIPA1L3 downstream targets either by unphysiological inhibition of Rap (if the truncated SIPA1L3 escapes degradation) or overactivation of Rap signaling (if the truncating variant results in loss-of-function). We hypothesize that this mechanism disturbs the complex embryonic lens development processes in the present family, leading to early cataract formation. Further functional studies are however necessary to elucidate the exact function of SIPA1L3 and its protein–protein interactions in the human lens.
In summary, we could map an interval on chromosome 19p13.11–q13.2 for autosomal recessive CC and identified a homozygous nonsense variant (c.4489C>T; p.(R1497*)) in the gene SIPA1L3. There is convincing evidence, although no definite proof, that this variant is causative for the cataract in the present family. This is the first sequence variant in a GAP associated with human cataract formation. Further characterization of SIPA1L3 function, its putative interaction partners and its role in lens development, and mutation screening of SIPA1L3 in additional cataract patients should provide insight into pathogenetic mechanism leading to cataract formation.
References
Reddy MA, Francis PJ, Berry V, Bhattacharya SS, Moore AT : Molecular genetic basis of inherited cataract and associated phenotypes. Surv Ophthalmol 2004; 49: 300–315.
Hansen L, Mikkelsen A, Nurnberg P et al: Comprehensive mutational screening in a cohort of Danish families with hereditary congenital cataract. Invest Ophthalmol Vis Sci 2009; 50: 3291–3303.
Berry V, Francis P, Reddy MA et al: Alpha-B crystallin gene (CRYAB) mutation causes dominant congenital posterior polar cataract in humans. Am J Hum Genet 2001; 69: 1141–1145.
Billingsley G, Santhiya ST, Paterson AD et al: CRYBA4, a novel human cataract gene, is also involved in microphthalmia. Am J Hum Genet 2006; 79: 702–709.
Heon E, Priston M, Schorderet DF et al: The gamma-crystallins and human cataracts: a puzzle made clearer. Am J Hum Genet 1999; 65: 1261–1267.
Kannabiran C, Rogan PK, Olmos L et al: Autosomal dominant zonular cataract with sutural opacities is associated with a splice mutation in the betaA3/A1-crystallin gene. Mol Vis 1998; 4: 21.
Litt M, Carrero-Valenzuela R, LaMorticella DM et al: Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human beta-crystallin gene CRYBB2. Hum Mol Genet 1997; 6: 665–668.
Litt M, Kramer P, LaMorticella DM, Murphey W, Lovrien EW, Weleber RG : Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA. Hum Mol Genet 1998; 7: 471–474.
Mackay DS, Boskovska OB, Knopf HL, Lampi KJ, Shiels A : A nonsense mutation in CRYBB1 associated with autosomal dominant cataract linked to human chromosome 22q. Am J Hum Genet 2002; 71: 1216–1221.
Riazuddin SA, Yasmeen A, Yao W et al: Mutations in betaB3-crystallin associated with autosomal recessive cataract in two Pakistani families. Invest Ophthalmol Vis Sci 2005; 46: 2100–2106.
Stephan DA, Gillanders E, Vanderveen D et al: Progressive juvenile-onset punctate cataracts caused by mutation of the gammaD-crystallin gene. Proc Natl Acad Sci USA 1999; 96: 1008–1012.
Sun H, Ma Z, Li Y et al: Gamma-S crystallin gene (CRYGS) mutation causes dominant progressive cortical cataract in humans. J Med Genet 2005; 42: 706–710.
Berry V, Francis P, Kaushal S, Moore A, Bhattacharya S : Missense mutations in MIP underlie autosomal dominant 'polymorphic' and lamellar cataracts linked to 12q. Nat Genet 2000; 25: 15–17.
Jamieson RV, Farrar N, Stewart K : Characterization of a familial t(16;22) balanced translocation associated with congenital cataract leads to identification of a novel gene, TMEM114, expressed in the lens and disrupted by the translocation. Hum Mutat 2007; 28: 968–977.
Pras E, Levy-Nissenbaum E, Bakhan T et al: A missense mutation in the LIM2 gene is associated with autosomal recessive presenile cataract in an inbred Iraqi Jewish family. Am J Hum Genet 2002; 70: 1363–1367.
Shiels A, Bennett TM, Knopf HL et al: The EPHA2 gene is associated with cataracts linked to chromosome 1p. Mol Vis 2008; 14: 2042–2055.
Zhang T, Hua R, Xiao W et al: Mutations of the EPHA2 receptor tyrosine kinase gene cause autosomal dominant congenital cataract. Hum Mutat 2009; 30: E603–E611.
Shiels A, Bennett TM, Knopf HL et al: CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q. Am J Hum Genet 2007; 81: 596–606.
Conley YP, Erturk D, Keverline A et al: A juvenile-onset, progressive cataract locus on chromosome 3q21-q22 is associated with a missense mutation in the beaded filament structural protein-2. Am J Hum Genet 2000; 66: 1426–1431.
Muller M, Bhattacharya SS, Moore T et al: Dominant cataract formation in association with a vimentin assembly disrupting mutation. Hum Mol Genet 2009; 18: 1052–1057.
Ramachandran RD, Perumalsamy V, Hejtmancik JF : Autosomal recessive juvenile onset cataract associated with mutation in BFSP1. Hum Genet 2007; 121: 475–482.
Azuma N, Yamaguchi Y, Handa H, Hayakawa M, Kanai A, Yamada M : Missense mutation in the alternative splice region of the PAX6 gene in eye anomalies. Am J Hum Genet 1999; 65: 656–663.
Jamieson RV, Perveen R, Kerr B et al: Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum Mol Genet 2002; 11: 33–42.
Semina EV, Ferrell RE, Mintz-Hittner HA et al: A novel homeobox gene PITX3 is mutated in families with autosomal-dominant cataracts and ASMD. Nat Genet 1998; 19: 167–170.
Aldahmesh MA, Khan AO, Mohamed JY, Alghamdi MH, Alkuraya FS : Identification of a truncation mutation of acylglycerol kinase (AGK) gene in a novel autosomal recessive cataract locus. Hum Mutat 2012; 33: 960–962.
Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Ruschendorf F, Nurnberg P : ALOHOMORA: a tool for linkage analysis using 10 K SNP array data. Bioinformatics 2005; 21: 2123–2125.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin -rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Li H, Durbin R : Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
Li H, Handsaker B, Wysoker A et al: The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Rimmer A, Phan H, Mathieson I et al: Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet 2014; 46: 912–918.
Wang K, Li M, Hakonarson H : ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
Abecasis GR, Auton A, Brooks LD et al: An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65.
Chomczynski P, Sacchi N : Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987; 162: 156–159.
Sherry ST, Ward MH, Kholodov M et al: dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001; 29: 308–311.
Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL http://evs.gs.washington.edu/EVS/ (accessed August 2014).
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D : MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–576.
Kumar P, Henikoff S, Ng PC : Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081.
Abecasis GR, Altshuler D, Auton A et al: A map of human genome variation from population-scale sequencing. Nature 2010; 467: 1061–1073.
Lachke SA, Ho JW, Kryukov GV et al: iSyTE: integrated Systems Tool for Eye gene discovery. Invest Ophthalmol Vis Sci 2012; 53: 1617–1627.
Bateman JB, Richter L, Flodman P et al: A new locus for autosomal dominant cataract on chromosome 19: linkage analyses and screening of candidate genes. Invest Ophthalmol Vis Sci 2006; 47: 3441–3449.
Kurachi H, Wada Y, Tsukamoto N et al: Human SPA-1 gene product selectively expressed in lymphoid tissues is a specific GTPase-activating protein for Rap1 and Rap2. Segregate expression profiles from a rap1GAP gene product. J Biol Chem 1997; 272: 28081–28088.
Singh L, Gao Q, Kumar A et al: The high-risk human papillomavirus type 16 E6 counters the GAP function of E6TP1 toward small Rap G proteins. J Virol 2003; 77: 1614–1620.
Rao PV, Zigler JS, Garland D : Analysis of small GTP-binding proteins of the lens by GTP overlay assay reveals the presence of unique GTP-binding proteins associated with fiber cells. Exp Eye Res 1997; 64: 219–227.
Walker J, Menko AS : Integrins in lens development and disease. Exp Eye Res 2009; 88: 216–225.
Kramer A, Green J, Pollard J, Tugendreich S : Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014; 30: 523–530.
Yao Z, Yu H, Xuan D, Sha Q, Hu J, Zhang J : Analysis of protein-protein interactions and proteomic profiles of normal human lenses. Curr Eye Res 2010; 35: 605–619.
Lichtstein D, McGowan MH, Russell P : Carper DA: Digitalis and digitalislike compounds down-regulate gene expression of the intracellular signaling protein 14-3-3 in rat lens. Hypertens Res 2000; 23 Suppl: S51–S53.
Noda Y, Horikawa S, Furukawa T et al: Aquaporin-2 trafficking is regulated by PDZ-domain containing protein SPA-1. FEBS Lett 2004; 568: 139–145.
Richter M, Murai KK, Bourgin C, Pak DT, Pasquale EB : The EphA4 receptor regulates neuronal morphology through SPAR-mediated inactivation of Rap GTPases. J Neurosci 2007; 27: 14205–14215.
Tsai IC, Amack JD, Gao ZH, Band V, Yost HJ, Virshup DM : A Wnt-CKIepsilon-Rap1pathway regulates gastrulation by modulating SIPA1L1, a Rap GTPase activating protein. Dev Cell 2007; 12: 335–347.
Reis LM, Tyler RC, Muheisen S et al: Whole exome sequencing in dominant cataract identifies a new causative factor, CRYBA2, and a variety of novel alleles in known genes. Hum Genet 2013; 132: 761–770.
Graw J, Klopp N, Illig T, Preising MN, Lorenz B : Congenital cataract and macular hypoplasia in humans associated with a de novo mutation in CRYAA and compound heterozygous mutations in P. Graefes Arch Clin Exp Ophthalmol 2006; 244: 912–919.
Pras E, Raz J, Yahalom V et al: A nonsense mutation (W9X) in CRYAA causes autosomal recessive cataract in an inbred Jewish Persian family. Invest Ophthalmol Vis Sci 2000; 41: 3511–3515.
Vicart P, Caron A, Guicheney P et al: A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet 1998; 20: 92–95.
Inagaki N, Hayashi T, Arimura T et al: Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem Biophys Res Commun 2006; 342: 379–386.
Safieh LA, Khan AO, Alkuraya FS et al: Identification of a novel CRYAB mutation associated with autosomal recessive juvenile cataract in a Saudi family. Mol Vis 2009; 15: 980–984.
Zhou G, Zhou N, Hu S, Zhao L, Zhang C, Qi Y : A missense mutation in CRYBA4 associated with congenital cataract and microcornea. Mol Vis 2010; 16: 1019–1024.
Cohen D, Bar-Yosef U, Levy J et al: Homozygous CRYBB1 deletion mutation underlies autosomal recessive congenital cataract. Invest Ophthalmol Vis Sci 2007; 48: 2208–2213.
Willoughby CE, Shafiq A, Ferrini W et al: CRYBB1 mutation associated with congenital cataract and microcornea. Mol Vis 2005; 11: 587–593.
Wang J, Ma X, Gu F et al: A missense mutation S228P in the CRYBB1 gene causes autosomal dominant congenital cataract. Chin Med J (Engl) 2007; 120: 820–824.
Gonzalez-Huerta LM, Messina-Baas OM, Cuevas-Covarrubias SA : A family with autosomal dominant primary congenital cataract associated with a CRYGC mutation: evidence of clinical heterogeneity. Mol Vis 2007; 13: 1333–1338.
Hansen L, Yao W, Eiberg H et al: Genetic heterogeneity in microcornea-cataract: five novel mutations in CRYAA, CRYGD, and GJA8. Invest Ophthalmol Vis Sci 2007; 48: 3937–3944.
Kaul H, Riazuddin SA, Shahid M et al: Autosomal recessive congenital cataract linked to EPHA2 in a consanguineous Pakistani family. Mol Vis 2010; 16: 511–517.
Mackay D, Ionides A, Kibar Z et al: Connexin46 mutations in autosomal dominant congenital cataract. Am J Hum Genet 1999; 64: 1357–1364.
Shiels A, Mackay D, Ionides A, Berry V, Moore A, Bhattacharya S : A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant "zonular pulverulent" cataract, on chromosome 1q. Am J Hum Genet 1998; 62: 526–532.
Ponnam SP, Ramesha K, Tejwani S, Ramamurthy B, Kannabiran C : Mutation of the gap junction protein alpha 8 (GJA8) gene causes autosomal recessive cataract. J Med Genet 2007; 44: e85.
Berry V, Gregory-Evans C, Emmett W et al: Wolfram gene (WFS1) mutation causes autosomal dominant congenital nuclear cataract in humans. Eur J Hum Genet 2013; 21: 1356–1360.
Jakobs PM, Hess JF, FitzGerald PG, Kramer P, Weleber RG, Litt M : Autosomal-dominant congenital cataract associated with a deletion mutation in the human beaded filament protein gene BFSP2. Am J Hum Genet 2000; 66: 1432–1436.
Aldahmesh MA, Khan AO, Mohamed J, Alkuraya FS : Novel recessive BFSP2 and PITX3 mutations: insights into mutational mechanisms from consanguineous populations. Genet Med 2011; 13: 978–981.
Bu L, Jin Y, Shi Y et al: Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract. Nat Genet 2002; 31: 276–278.
Smaoui N, Beltaief O, BenHamed S et al: A homozygous splice mutation in the HSF4 gene is associated with an autosomal recessive congenital cataract. Invest Ophthalmol Vis Sci 2004; 45: 2716–2721.
Bidinost C, Matsumoto M, Chung D et al: Heterozygous and homozygous mutations in PITX3 in a large Lebanese family with posterior polar cataracts and neurodevelopmental abnormalities. Invest Ophthalmol Vis Sci 2006; 47: 1274–1280.
Berry V, Francis PJ, Prescott Q, Waseem NH, Moore AT, Bhattacharya SS : A novel 1-bp deletion in PITX3 causing congenital posterior polar cataract. Mol Vis 2011; 17: 1249–1253.
Lachke SA, Alkuraya FS, Kneeland SC et al: Mutations in the RNA granule component TDRD7 cause cataract and glaucoma. Science 2011; 331: 1571–1576.
Chen J, Ma Z, Jiao X et al: Mutations in FYCO1 cause autosomal-recessive congenital cataracts. Am J Hum Genet 2011; 88: 827–838.
Pras E, Frydman M, Levy-Nissenbaum E et al: A nonsense mutation in the glucosaminyl (N-acetyl) transferase 2 gene (GCNT2): association with autosomal recessive congenital cataracts. Invest Ophthalmol Vis Sci 2004; 45: 1940–1945.
Mayr JA, Haack TB, Graf E et al: Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome. Am J Hum Genet 2012; 90: 314–320.
Hansen L, Comyn S, Mang Y et al: The myosin chaperone UNC45B is involved in lens development and autosomal dominant juvenile cataract. Eur J Hum Genet 2014; 22: 1290–1297.
Acknowledgements
We are grateful to the patients and their parents for their participation in the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Evers, C., Paramasivam, N., Hinderhofer, K. et al. SIPA1L3 identified by linkage analysis and whole-exome sequencing as a novel gene for autosomal recessive congenital cataract. Eur J Hum Genet 23, 1627–1633 (2015). https://doi.org/10.1038/ejhg.2015.46
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.46
This article is cited by
-
Autosomal recessive cataract (CTRCT18) in the Yakut population isolate of Eastern Siberia: a novel founder variant in the FYCO1 gene
European Journal of Human Genetics (2021)
-
Genetic landscape of isolated pediatric cataracts: extreme heterogeneity and variable inheritance patterns within genes
Human Genetics (2019)
-
RNA sequencing-based transcriptomic profiles of embryonic lens development for cataract gene discovery
Human Genetics (2018)
-
A 16.7 kb deletion in Sipa1l3 is associated with juvenile cataract in mice
Mammalian Genome (2017)
-
Comparative expression study of sipa family members during early Xenopus laevis development
Development Genes and Evolution (2016)
