
Abstract
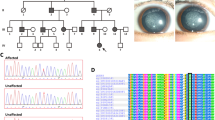
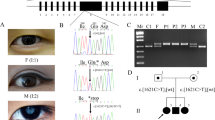
Congenital acorea is a rare disease with the absence of a pupil in the eye. To date, only one family and two isolated cases with congenital acorea have been reported. The gene associated with acorea has not been identified. In this study, we recruited a Chinese family acorea-microphthalmia-cataract syndrome. By analyzing the whole-exome sequencing (WES) data of this Chinese family, we revealed the association of a novel heterozygous variant, NM_005267.5:c.137G>A (p.G46E) in the gap junction protein alpha 8 (GJA8) gene encoding connexin 50 or CX50, with familial acorea-microphthalmia-cataract syndrome. Additionally, another variant, NM_005267.5:c.151G>A (p.D51N) in GJA8, was identified to co-segregate with this syndrome in an unrelated Japanese family. Ectopic expression of p.G46E and p.D51N mutant GJA8 genes in cultured cells caused protein mislocalization, suggesting that the p.G46E and p.D51N mutations in GJA8 impaired the function of the gap junction channels. These results established GJA8 as the first gene associated with familial acorea-microphthalmia-cataract syndrome.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

Data availability
The genetic variants of GJA8 identified in this study have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). The accession numbers for G46E and D51N are SCV003920679 and SCV004011733, respectively. All other data are available within the article or upon reasonable request.
References
Graw J. Eye development. Curr Top Dev Biol. 2010;90:343–86.
Shah SP, Taylor AE, Sowden JC, Ragge NK, Russell-Eggitt I, Rahi JS, et al. Anophthalmos, microphthalmos, and typical coloboma in the United Kingdom: a prospective study of incidence and risk. Investig Ophthalmol Vis Sci. 2011;52:558–64.
Richardson R, Sowden J, Gerth-Kahlert C, Moore AT, Moosajee M. Clinical utility gene card for: non-syndromic microphthalmia including next-generation sequencing-based approaches. Eur J Hum Genet. 2017;25:512.
Reis LM, Semina EV. Conserved genetic pathways associated with microphthalmia, anophthalmia, and coloboma. Birth Defects Res C Embryo Today. 2015;105:96–113.
Ceroni, Aguilera-Garcia F, Chassaing D, Bax DA N, Blanco-Kelly F, Ramos P, et al. New GJA8 variants and phenotypes highlight its critical role in a broad spectrum of eye anomalies. Hum Genet. 2019;138:1027–42.
Gillespie RL, O’Sullivan J, Ashworth J, Bhaskar S, Williams S, Biswas S, et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology. 2014;121:2124–37. e2121-2122.
Koval M. Pathways and control of connexin oligomerization. Trends Cell Biol. 2006;16:159–66.
Pfenniger A, Wohlwend A, Kwak BR. Mutations in connexin genes and disease. Eur J Clin Investig. 2011;41:103–16.
Rouillac C, Roche O, Marchant D, Bachner L, Kobetz A, Toulemont PJ, et al. Mapping of a congenital microcoria locus to 13q31-q32. Am J Hum Genet. 1998;62:1117–22.
Tawara A, Inomata H. Familial cases of congenital microcoria associated with late onset congenital glaucoma and goniodysgenesis. Jpn J Ophthalmol. 1983;27:63–72.
Ramprasad VL, Sripriya S, Ronnie G, Nancarrow D, Saxena S, Hemamalini A, et al. Genetic homogeneity for inherited congenital microcoria loci in an Asian Indian pedigree. Mol Vis. 2005;11:934–40.
Fares-Taie L, Gerber S, Tawara A, Ramirez-Miranda A, Douet JY, Verdin H, et al. Submicroscopic deletions at 13q32.1 cause congenital microcoria. Am J Hum Genet. 2015;96:631–9.
Kondo H, Tahira T, Yamamoto K, Tawara A. Familial acorea, microphthalmia and cataract syndrome. Br J Ophthalmol. 2013;97:1155–60.
Ramasubramanian S, Majumder PD. Acorea: a rare congenital anomaly. Indian J Ophthalmol. 2018;66:450.
Priyambada P, Prabu RV, Wasnik RB, Ranjini H. A rare case of acorea: congenital absence of pupil. J Clin Ophthalmol Res. 2021;9:85–87.
Wu X, Long E, Lin H, Liu Y. Prevalence and epidemiological characteristics of congenital cataract: a systematic review and meta-analysis. Sci Rep. 2016;6:28564.
Shiels A, Mackay D, Ionides A, Berry V, Moore A, Bhattacharya S. A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant “zonular pulverulent” cataract, on chromosome 1q. Am J Hum Genet. 1998;62:526–32.
Ma AS, Grigg JR, Ho G, Prokudin I, Farnsworth E, Holman K, et al. Sporadic and familial congenital cataracts: mutational spectrum and new diagnoses using next-generation sequencing. Hum Mutat. 2016;37:371–84.
Maeda S, Tsukihara T. Structure of the gap junction channel and its implications for its biological functions. Cell Mol Life Sci. 2011;68:1115–29.
Myers JB, Haddad BG, O’Neill SE, Chorev DS, Yoshioka CC, Robinson CV, et al. Structure of native lens connexin 46/50 intercellular channels by cryo-EM. Nature. 2018;564:372–7.
Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, et al. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009;458:597–602.
Tong JJ, Minogue PJ, Guo W, Chen TL, Beyer EC, Berthoud VM, et al. Different consequences of cataract-associated mutations at adjacent positions in the first extracellular boundary of connexin50. Am J Physiol Cell Physiol. 2011;300:C1055–1064.
Sun W, Xiao X, Li S, Guo X, Zhang Q. Mutational screening of six genes in Chinese patients with congenital cataract and microcornea. Mol Vis. 2011;17:1508–13.
Fan F, Luo Y, Wu J, Gao C, Liu X, Mei H, et al. The mutation spectrum in familial versus sporadic congenital cataract based on next-generation sequencing. BMC Ophthalmol. 2020;20:361.
Azuma N, Hirakiyama A, Inoue T, Asaka A, Yamada M. Mutations of a human homologue of the Drosophila eyes absent gene (EYA1) detected in patients with congenital cataracts and ocular anterior segment anomalies. Hum Mol Genet. 2000;9:363–6.
Reis LM, Tyler RC, Muheisen S, Raggio V, Salviati L, Han DP, et al. Whole exome sequencing in dominant cataract identifies a new causative factor, CRYBA2, and a variety of novel alleles in known genes. Hum Genet. 2013;132:761–70.
Gonzalez-Huerta LM, Messina-Baas OM, Cuevas-Covarrubias SA. A family with autosomal dominant primary congenital cataract associated with a CRYGC mutation: evidence of clinical heterogeneity. Mol Vis. 2007;13:1333–8.
Ganatra S, Kekunnaya R, Sachdeva V. Bilateral congenital membranous cataracts due to Glucosaminyl (N-Acetyl) Transferase 2 (GCNT2) mutation: Life-saving genetic analysis. Indian J Ophthalmol. 2022;70:2622–3.
Church RL, Wang JH, Steele E. The human lens intrinsic membrane protein MP70 (Cx50) gene: clonal analysis and chromosome mapping. Curr Eye Res. 1995;14:979–81.
Kumar NM, Gilula NB. Molecular biology and genetics of gap junction channels. Semin Cell Biol. 1992;3:3–16.
Rong P, Wang X, Niesman I, Wu Y, Benedetti LE, Dunia I, et al. Disruption of Gja8 (alpha8 connexin) in mice leads to microphthalmia associated with retardation of lens growth and lens fiber maturation. Development. 2002;129:167–74.
Arora A, Minogue PJ, Liu X, Addison PK, Russel-Eggitt I, Webster AR, et al. A novel connexin50 mutation associated with congenital nuclear pulverulent cataracts. J Med Genet. 2008;45:155–60.
Banks EA, Toloue MM, Shi Q, Zhou ZJ, Liu J, Nicholson BJ, et al. Connexin mutation that causes dominant congenital cataracts inhibits gap junctions, but not hemichannels, in a dominant negative manner. J Cell Sci. 2009;122:378–88.
Rubinos C, Villone K, Mhaske PV, White TW, Srinivas M. Functional effects of Cx50 mutations associated with congenital cataracts. Am J Physiol Cell Physiol. 2014;306:C212–220.
Tong X, Aoyama H, Tsukihara T, Bai D. Charge at the 46th residue of connexin 50 is crucial for the gap-junctional unitary conductance and transjunctional voltage-dependent gating. J Physiol. 2014;592:5187–202.
Mese G, Sellitto C, Li L, Wang HZ, Valiunas V, Richard G, et al. The Cx26-G45E mutation displays increased hemichannel activity in a mouse model of the lethal form of keratitis-ichthyosis-deafness syndrome. Mol Biol Cell. 2011;22:4776–86.
Kronengold J, Trexler EB, Bukauskas FF, Bargiello TA, Verselis VK. Pore-lining residues identified by single channel SCAM studies in Cx46 hemichannels. Cell Commun Adhes. 2003;10:193–9.
Sanchez HA, Villone K, Srinivas M, Verselis VK. The D50N mutation and syndromic deafness: altered Cx26 hemichannel properties caused by effects on the pore and intersubunit interactions. J Gen Physiol. 2013;142:3–22.
Lopez W, Ramachandran J, Alsamarah A, Luo Y, Harris AL, Contreras JE. Mechanism of gating by calcium in connexin hemichannels. Proc Natl Acad Sci USA. 2016;113:E7986–E7995.
Wang L, Zou T, Lin Y, Li L, Zhang P, Gong B, et al. Identification of a novel homozygous variant in the CNGA1 gene in a Chinese family with autosomal recessive retinitis pigmentosa. Mol Med Rep. 2020;22:2516–20.
Acknowledgements
The authors thank all the participants in this study.
Funding
The National Natural Science Foundation of China (nos. 81770935 (HZ), 82371059 (HZ), 81800830 (SD)), the grant from the Department of Science and Technology of Sichuan Province, China (nos. 2023JDZH0002 (HZ), 2022JDTD0024 (BG)), the Chengdu Science and Technology Bureau (no. 2022-YF05-01984-SN (HZ)), Young and Middle-aged Health Science and Technology Innovation Talent Training Project (Outstanding Young Persons) of Henan Province (no. YXKC2022025) (SD), Medical Science and Technology Project (the Key Project Jointly Built by the Province and the Ministry) of Henan Province (no. SBGJ202102167, (SD)), and the Key Research and Development and Promotion Project (Science and Technology) program of Henan Province (no. 192102310077 (SD)).
Author information
Authors and Affiliations
Contributions
SD, WZ, and QL recruited the Chinese participants. TZ, FZ, TW, and JW performed sequencing, vector construction, and cell culture, and analyzed the data. TN, IM, HK recruited the Japanese participants, performed the experiments and analyzed the data. SD, GB, QL, and HZ designed the experiments, and analyzed the data. QL and HZ supervised the project. TZ and HZ wrote the manuscript.
Corresponding authors
Ethics declarations
Ethical
This study complied with the requirements of the Declaration of Helsinki, and was approved by ethics committees of all participating institutions, including the First Affiliated Hospital of Zhengzhou University, China; Sichuan Provincial People’s Hospital, China; and the University of Occupational and Environmental Health, Japan. Written informed consent for publication of details and images was obtained from all the participants or their guardians.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dong, S., Zou, T., Zhen, F. et al. Association of variants in GJA8 with familial acorea-microphthalmia-cataract syndrome. Eur J Hum Genet 32, 413–420 (2024). https://doi.org/10.1038/s41431-023-01503-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-023-01503-9
This article is cited by
-
Artificial intelligence â the next generation of sequencing?
European Journal of Human Genetics (2024)
