
Abstract
Consanguineous marriages have been widely practiced in several global communities with varying rates depending on religion, culture, and geography. In consanguineous marriages, parents pass to their children autozygous segments known as homozygous by descent segments. In this study, single-nucleotide polymorphisms were analyzed in 165 unrelated Lebanese people from Greek Orthodox, Maronite, Shiite and Sunni communities. Runs of homozygosity, total inbreeding levels, remote consanguinity, and population admixture and structure were estimated. The inbreeding coefficient value was estimated to be 1.61% in offspring of unrelated parents over three generations and 8.33% in offspring of first cousins. From these values, remote consanguinity values, resulting from genetic drift or recurrent consanguineous unions, were estimated in offspring of unrelated and first-cousin parents to be 0.61 and 1.2%, respectively. This remote consanguinity value suggests that for any unrelated marriages in Lebanon, the mates could be related as third cousins or as second cousins once removed. Under the assumption that 25% of marriages occur between first cousins, the mean inbreeding value of 2.3% may explain the increased incidence of recessive disease in offspring. Our analysis reveals a common ancestral population in the four Lebanese communities we studied.
Similar content being viewed by others


Introduction
Consanguineous marriages encouraged by socio-cultural factors are widely practiced around the world, particularly in the Middle East.1, 2 First cousin unions, comprising 20–30% of all marriages, are the most common form of consanguinity.1, 2, 3 In isolates or small isolated populations, the genetic drift and the choice of mates are responsible for the ‘remote’ consanguinity (RC) and the ‘apparent’ or ‘immediate’ consanguinity,4 respectively, which are combined within the mean inbreeding coefficient (F).4
Currently, both mathematical and biological methods are used to estimate the F in a population. A mathematical calculation using the Malécot formula applied to genealogies, as well as consanguineous marriage statistics from previous studies, led to an F-value of 1.56% for a Lebanese population.3, 7, 8, 9 Using biological methodologies, an F estimation was first calculated using a microsatellite panel and FEstim algorithm and was then improved using genome-wide single-nucleotide polymorphism (SNP) chip-based advanced algorithms.5, 6 These estimations were based on the genotyping of a large number of genetic markers to infer the individual genomic proportion that is homozygous by descent (HBD). Two parameters were then calculated; the proportion of genomic HBD and the average length of the HBD segments, which are the indicators of the inbreeding and the age of common ancestors, respectively.5, 7
Herein, we report an alternative way to measure genomic homozygosity (GH), through the counting of runs of homozygosity (ROH), to calculate HBD and subsequently to estimate RC. This ROH approach does not require SNP frequencies, which are essential for the FEstim approach. Therefore, it is of interest to compare HBD estimates made using these two genome-wide approaches.
The ROH approach was applied to samples stratified under the criteria of consanguinity and religious status because the Lebanese population is divided into different religious communities within which numerous consanguineous marriages occur.8 This approach was carried out to estimate and compare ROH profiles, HBD, RC values and evaluate possible common ancestry.
Materials and methods
Subjects and comparative datasets
A total of 165 DNA samples were stratified into four subpopulations based on religion: 72 Christians (25 Greek Orthodox (GO) and 47 Maronite (MA)) and 93 Muslims (55 Shiite (SH) and 38 Sunni (SU)). Subjects in each of the four groups were further subdivided into two classes according to whether their parents were first cousins (FCO) (53 samples) or unrelated (URO) (112 samples) (Table 1).
Approval to conduct the study was obtained from the Ethics Committee of Saint-Joseph University-Lebanon and the French State administration (CNIL: Commission Nationale Informatique et Libertés). Informed consent was obtained from all participants. DNA was extracted from lymphocytes using standard methods.9
Comparative data (132 samples) were obtained from panmictic samples extracted from the HapMap 3 consortium data, the CEU (northwest European derived population from Utah, USA) and the TSI (Tuscans from Italy) populations.10
A second comparative data set was obtained from the Human Genome Diversity Panel (HGDP-CEPH), containing 938 unrelated individuals originating from 51 global populations.11, 12, 13
DNA arrays
Genotyping was performed using the Affymetrix Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA, USA) (90 samples) and the Affymetrix Cytogenetic 2.7 M Whole-Genome Microarrays (Affymetrix) (75 samples) according to the manufacturer’s protocol.
The SNP6.0 array contains 1.8 M probes (906 600 SNPs and 946 000 non-polymorphic markers) and captures 82% of all HapMap 2 variations with r2≥0.8 in CEU samples.14 The Cyto 2.7 M contains 2.7 million non-polymorphic markers and 400 000 SNPs.
Using these two types of arrays was reasonable because they were found to be equally well suited to detect ROH.15 However, because these arrays differ in SNP density, they might differ in the total length of genomic ROH provided. To investigate this issue, nine DNA samples were simultaneously genotyped with both arrays. The total length of genomic ROH of the 75 Cyto 2.7 M samples differed by a fixed correction factor of 1.39 and 1.3 for FCO and URO, respectively. The data from the two types of arrays were then combined and analyzed together after adjustment with the corresponding correction factor.
ROH analyses and genomic homozygosity estimation
Chromosome Analysis Suite (ChAS) v1.0.1 (Affymetrix) and Affymetrix Genotyping Console (GTC4.0) (Affymetrix) were used for the analysis of the Cytogenetic 2.7 M and the Genome-Wide Human SNP Array 6.0 data, respectively. Loss of heterozygosity regions, extracted from both types of arrays, were considered as ROH and were analyzed with Microsoft Excel (Microsoft, Redmond, WA, USA). The sizes of the various ROH regions were calculated using the physical position of the SNPs at the start and at the end of each region. The individual ROH distributions were studied using two different analyses.
The first analysis was performed to compare ROH distributions between Lebanese communities and European populations (Figure 1). The average total length of the genomic ROH estimated only from SNP6.0 was organized into six classes based on size as described by Kirin et al16 because the Illumina 650Y (Illumina, San Diego, CA, USA) used by these authors contains as many SNPs as SNP6.0. In each size class, the average total length ROH was calculated for the URO or FCO within each religious subgroup.

Lebanese and European distribution of ROH. The average total length of genomic ROH classified by length is plotted for each Lebanese religious subpopulation versus the European group. In each length category, columns 2–5 and columns 6–9 represent URO and FCO samples, respectively.
The second analysis was performed to provide a better estimation of the observed GH within communities as well as between URO and FCO. The individual total ROH estimated either from the SNP6.0 or Cyto2.7M arrays were combined using the correction factor previously calculated.
For the calculation of the observed GH percentages in each individual, the sizes of the ROH regions greater than 1.5 Mb (excluding the sex chromosomes) were summed and then divided by the total autosomal length (2.867.766 kb for hg18).17 To avoid underestimation of the GH and the RC inflation of GH measurements, a 1.5 Mb threshold was defined. Indeed, McQuillan et al18 recommend that a≥1.5 Mb threshold be applied to ROH measurements for the identity by descent percentage estimation. They state that all ROH shorter than this 1.5 Mb cutoff reflect linkage disequilibrium patterns of ancient origin rather than the effects of more recent endogamy and parental relatedness.
To study the shared ROH regions between individuals and communities, the ROH regions generated from the SNP6.0 arrays were aligned using the custom tracks tool in the UCSC Genome Bioinformatics Site (http://genome.ucsc.edu). ROH regions were defined by an uninterrupted sequence of ≥50 homozygous SNP markers and a minimum size of 3 Mb. Shared regions between individuals from different religious groups were analyzed and classified by size and by community.
Homozygosity estimation using FEstim
For the 90 samples genotyped by Affymetrix SNP6.0 arrays, the inbreeding coefficients (F) were estimated using maximum likelihood with a hidden Markov model approach implemented in FEstim using submaps.5, 6
All genotyped individuals had call rates ≥95%. After quality control checks requiring SNP call rates≥95%, a Hardy–Weinberg P-value≤10−7 and SNP minor allele frequency of ≥5%, a total of 646 200 SNPs remained for FEstim analysis.
Total and remote consanguinity estimations
To take into account the baseline homozygosity present in every population, the GH value observed in the Lebanese samples was corrected by the baseline value (GHpp) estimated in 132 controls extracted from panmictic populations (HapMap 3). HBD was then calculated using the probability equation (1-GH)=(1-GHpp) (1-HBD).19
The RC of the URO samples was directly estimated from their HBD value, and the RC of the FCO samples was calculated by partitioning the HBD value following the (1-HBD)=(1-RC) (1-1/16) equation because only 1/16 of the genome is expected to be homozygous due to their parental kinship.
Means values for GH, HBD and RC in the FCO and URO samples were calculated by weighting the mean individual values of each religious community by their own relative frequency in the present population (GO: 1/12—MA: 3/12—SU: 4/12—SH: 4/12).
Population structure
Samples genotyped with Affymetrix SNP6.0 arrays were compared with those of 938 individuals from the HGDP-CEPH panel genotyped with Illumina650Y using principal component analysis (PCA). This analysis was performed using the software SmartPCA on 19 061 SNPs common to the datasets.20
Results
Identified ROH
Among the 90 samples analyzed with SNP6.0 array (55 URO, 35 FCO), a total of 772 ROH (21% in URO and 79% in FCO) were observed. ROHs lengths ranged from 3.01 to 52.57 Mb (7.54±6.57 Mb) in URO and from 3 to 57.64 Mb (12.13±10.13 Mb) in FCO, which is consistent with the inbred status of FCO.
Among the 55 URO individuals, a total of 161 ROHs were found >3 Mb in length with a minimum of 50 markers. The longest track identified in the 55 URO individuals was 52.57 Mb, which is much greater than the 27.32 and 17.91 Mb previously described by Li et al21 and Gibson et al,22 respectively. These long tracts of homozygosity were frequently observed in our cohort and are due to RC resulting from consanguineous marriages going back more than three generations.
Distribution of ROH regions between Lebanese communities and comparison with European populations
The average total length of genome ROH in each size category showed no significant difference between the URO samples in the four Lebanese communities (Figure 1). This demonstrates no genomic difference between these four subpopulations in the distribution of ROH.
The lack of difference in the distribution of ROH reflects the ‘memory’ of demographic and genetic history in a population. Our observation is consistent with the supposed common anthropological origin of all Lebanese, their demographic history, and their practice of marriage between relatives.
The observed ROH distribution in FCO was different between Christians and Muslims, (particularly 8–16 Mb ROH) indicating that first-cousin unions are mostly sporadic in Christian communities but recurrent in Muslim communities (Figure 1).
The distribution of Lebanese ROHs>1 Mb was significantly more frequent than in the European populations (Figure 1). This indicates a high level of homozygosity resulting from marriages between relatives that frequently occur in Lebanon, but that rarely occur in Europe.16
This ROH comparison showed that a moderate proportion of homozygosity (ROH 2–4 Mb) corresponds to an ancient parental relatedness as a result of genetic drift or recurrent consanguineous unions. Consequently, the largest fraction of GH observed in URO as well as in FCO (ROH >8 Mb) corresponds to recent parental relatedness going back more than three generations (Figure 1).
Genomic homozygosity, total and remote consanguinity in URO
Observed GH percentages were calculated from both types of arrays and combined using each array correction factor value (Table 1).
The individual GH means for URO in the four communities were nearly identical. The weighted mean for the whole population was found to be 1.61%, a value similar to the observed means in endogamous Dalmatians and Orcadians (1.3 and 1.1%, respectively).18
The baseline GH GHpp value was measured in outbred panmictic populations (CEU and TSI). This observed value of GHpp (1%) allowed us to then correct the observed GH values in the Lebanese population to infer the HBD and the RC values in URO (Table 1). The RC value observed in Lebanese URO, was roughly equal to 0.61%, corresponding to ~1/163. This value suggests that for any unrelated marriages in Lebanon, the mates could be related as third cousins (1/256) or second cousins once removed (1/128).
Genomic homozygosity, total and remote consanguinity in FCO
HBD values are expected to be higher than 6.25% within FCO due to RC, but this was observed only in the Muslim communities (Table 1). Indeed, higher RC values were observed within Muslim FCO (1.3 and 2.3%) when compared with those of URO (0.6 and 0.5%). In addition, RC values were not observed in Christian communities (Table 1). The difference observed between Muslims and Christians was not due to low sample size because RC was similar within URO in the four communities.
These findings are consistent with the fact that first-cousin unions are mostly sporadic in Christian communities but are recurrent in Muslim communities. Therefore, in Christian communities, the RC value within FCO samples was masked by the larger variance of HBD within FCO samples when compared with URO. On the contrary, due to recurrent unions between relatives in Muslim families, significantly higher RC values in FCO than in URO were expected.
Genomic homozygosity, total and remote consanguinity in the whole population
Mean weighted values for GH, HBD and RC in URO and FCO allowed us to compare our observed results with previously published data (Table 1). Under the assumption that 25% of the unions in Lebanon are between first cousins, HBD values (Table 1) lead to an estimated F mean value equal to 2.3%, a much higher percentage than the previously estimated (1.56%).3, 23, 24, 25
Homozygosity estimation using FEstim
Among the 90 individuals, 48 had an inbreeding coefficient F significantly different than zero (Figure 2). In fact, we found that 33 of 34 FCO individuals and 14 of 55 (25%) URO individuals were inbred. Among these inbred individuals, FCO had, on average, a higher F-value when compared with URO, but there was some overlap in the lower values (0.7–15.8% vs 0.6–7.5%, respectively). URO individuals identified as inbred were present in all communities (four SH, four MA, three GO, and three SU). The lowest and the highest F-values for FCO were found in the GO and SH communities, respectively.

FEstim inbreeding coefficient estimates for the 90 Lebanese samples, composed of 35 first cousin’s offspring (FCO) and 55 offspring of unrelated parents (URO). Each community is represented by a different color: GO: Greek Orthodox, MA: Maronite, SH: Shiite, SU: Sunni. The blue line represents the limit of F significantly different from zero.
The mean F-values, weighted by communities, were equal to 8.2 and 0.7% for FCO and URO, respectively (Table 1). Because FEstim relies on SNP frequencies, there is no need to correct the baseline GH. Thus, FEstim results are directly comparable to the HBD results of the ROH method (Table 1). For instance, the RC values for URO individuals (0.73%) can be directly compared with the value previously obtained from the ROH method (0.61%). Under the assumption that 25% of marriages occur between first cousins, the mean population inbreeding coefficient is 2.6% which is similar to but slightly higher than the estimate obtained from the ROH method (2.3%).
Shared individual ROH regions and population structure
Of the ROH regions shared by several individuals, individuals from the same community shared 9.6% of these regions and 90.4% were shared by individuals of two or more religious communities (Table 2). The mean size of the ROHs shared by two subpopulations was greater than that shared by three subpopulations, which was also greater than that shared by four subpopulations. These findings suggest that the ROHs identified in the four Lebanese subpopulations were inherited from a common ancestral population. Indeed, under the hypothesis that the size of the shared ROHs reflects its date of origin, partially inbred subpopulations known to be geographically and religiously isolated for centuries and resulting in recurrent crossing-over explain the fact that the mean size of the overlapping ROHs are inversely proportional to the number of communities sharing these blocks (Table 2).
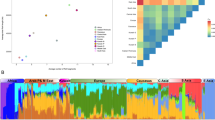
A PCA confirmed that all individuals were of Middle-Eastern origin (Figure 3a). Within the Middle Easter populations (Figure 3b), samples from the SU community overlapped the most with Palestinians from the central region of Israel and slightly with the Bedouins from the Negev region of Israel. Samples from the GO community slightly overlapped with the Druze of Northern Israel. Within Lebanon, no clear separation was detected between communities (Figure 3b), but overlaps within each community (Christian and Muslim) were found as noted by Haber et al.8 Such results are consistent with the common ancestry illustrated by the shared ROH previously mentioned.

Principal component analysis (PCA) of the Lebanese individuals (dark green) and the HGDP-CEPH panel individuals (a). PCA by communities with only the HGDP-CEPH panel Middle-Eastern individuals zoomed in on the Lebanese individuals (triangles) (b). GO: Greek Orthodox, MA: Maronite, SH: Shiite, SU: Sunni.
Discussion
Using genomic information, we studied inbreeding levels, RC, and population admixture within the Lebanese population.
The inaccuracy of genealogical data and the fact that the calculated value is only an expected quantity with respect to the genealogy is the rationale behind using genome-wide analysis. This allowed us to obtain a more accurate estimation of the level of inbreeding in the Lebanese population.6 In previous studies, F was estimated using Malécot’s formula applied to sociological data from first cousin unions. Based on the assumption that 25% of marriages occur between first cousins, F was found to be equal to 1.56%.22, 23, 24 Under the same assumption, the ROH method and FEstim estimated F equal to 2.3 and 2.6%, respectively; these values are significantly higher than what was previously found (1.56%). Therefore, these genome-wide findings lead to the inclusion of Lebanon in the group of Middle Eastern countries that show high levels of inbreeding.
The estimates of F showed marginal differences between the two genome-wide approaches. The FEstim approach relies on SNP frequencies that can be problematic to estimate, especially if the studied population size is small or if it is not well represented in reference panels. In this case, ROH would be a better approach because the ROH size threshold is well defined in the studied population.
The RC found within all URO in Lebanon is roughly equal to 0.61%, suggesting that for any ‘unrelated’ marriages in Lebanon the mates could actually be related as third cousins or as second cousins once removed. Moreover, subpopulation differences were observed with higher RC values detected among Muslims FCO, most likely due to preferential and recurrent FC marriages in some families. Among Christians, consanguineous unions are more sporadic, but Muslim communities were found to be heterogeneous with some subentities strongly inbred and others almost panmictic. Thus, our analyses of ROH, HBD, and RC in the Lebanese population indicate a more recent rather than ancient relatedness.
Because the RC value we found multiplies the risk of rare recessive diseases by 60 (allelic frequencies of 10−4 versus panmictic risk of 10−8), this explains the prevalence of such diseases in Lebanon, not only in offspring of related couples but also among offspring of unrelated couples.
With recessive diseases, calculating expected proportion of homozygous or compound heterozygous patients or frequencies of pathogenic alleles can now be performed using the Ten Kate et al equation using an accurate rate of F. For instance, compared with the previous F-value (1.56%), the present F-value (2.3%) increases the frequency of an autosomal recessive disease by 11.42%. Ranking the prevalence of autosomal recessive disorders will have social and clinical relevance as well as it will allow the setting up of priorities for genetic testing at the population level.26, 27
The presence of admixture in the current Lebanese subpopulations and PCA results inferred a genomic relationship. In fact, all Lebanese communities share similarities between each other and within the Middle-Eastern populations, regardless of religion status. This can be explained by the ancient history of the region despite geographical isolation and socio-cultural factors.
Recent studies have established guidelines that reduce the occurrence of false-positive and false-negative results in assigning parental relatedness of a proband on the basis of genomic testing that detects ROH.28, 29 In the present study, we provide new strategies that overcome those errors by stratifying the studied population into inbred status (URO vs FCO) and estimating RC by taking into account the average basic value of GH associated with various panmictic populations.
Change history
17 September 2015
This paper has been corrected since online publication and a corrigendum also appears in this issue
References
Bittles AH, Black ML : Evolution in health and medicine Sackler colloquium: consanguinity, human evolution, and complex diseases. Proc Natl Acad Sci USA 2010; 107: 1779–1786.
Alkuraya FS : Autozygome decoded. Genet Med 2010; 12: 765–771.
Hamamy H, Antonarakis SE, Cavalli-Sforza LL et al: Consanguineous marriages, pearls and perils: Geneva International Consanguinity Workshop Report. Genet Med Off J Am Coll Med Genet 2011; 13: 841–847.
Cavalli-Sforza LL, Bodmer WF The Genetics of Human Populations 1971.
Leutenegger AL, Prum B, Génin E et al: Estimation of the inbreeding coefficient through use of genomic data. Am J Hum Genet 2003; 73: 516–523.
Leutenegger AL, Sahbatou M, Gazal S et al: Consanguinity around the world: what do the genomic data of the HGDP-CEPH diversity panel tell us? Eur J Hum Genet 2011; 19: 583–587.
Browning SR, Browning BL : High-resolution detection of identity by descent in unrelated individuals. Am J Hum Genet 2010; 86: 526–539.
Haber M, Gauguier D, Youhanna S et al: Genome-wide diversity in the levant reveals recent structuring by culture. PLoS Genet 2013; 9: e1003316.
Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
International HapMap 3 Consortium. et al.. Integrating common and rare genetic variation in diverse human populations. Nature 2010; 467: 52–58.
Cann HM, de Toma C, Cazes L et al: A human genome diversity cell line panel. Science 2002; 296: 261–262.
Li JZ, Absher DM, Tang H et al: Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008; 319: 1100–1104.
Rosenberg NA : Standardized subsets of the HGDP-CEPH Human Genome Diversity Cell Line Panel, accounting for atypical and duplicated samples and pairs of close relatives. Ann Hum Genet 2006; 70: 841–847.
International HapMap Consortium International HapMap Consortium, Frazer KA International HapMap Consortium, Ballinger DG International HapMap Consortium, Cox DR et al: A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449: 851–861.
Bødker JS, Gyrup C, Johansen P et al: Performance comparison of Affymetrix SNP6.0 and cytogenetic 2.7M whole-genome microarrays in complex cancer samples. Cytogenet Genome Res 2013; 139: 80–87.
Kirin M, McQuillan R, Franklin CS et al: Genomic runs of homozygosity record population history and consanguinity. PLoS One 2010; 5: e13996.
Kearney HM, Kearney JB, Conlin LK : Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin Lab Med 2011; 31: 595–613.
McQuillan R, Leutenegger AL, Abdel-Rahman R et al: Runs of homozygosity in European populations. Am J Hum Genet 2008; 83: 359–372.
Wright S. : Evolution and the Genetics of Populations; Vol. 2. The Theory of Gene Frequencies. Chicago: University of Chicago Press, 1968.
Patterson N, Price AL, Reich D : Population structure and eigenanalysis. PLoS Genet 2006; 2: 2074–2093.
Li LH, Ho SF, Chen CH et al: Long contiguous stretches of homozygosity in the human genome. Hum Mutat 2006; 27: 1115–1121.
Gibson J, Morton NE, Collins A : Extended tracts of homozygosity in outbred human populations. Hum Mol Genet 2006; 15: 789–795.
Khlat M : Consanguineous marriage and reproduction in Beirut, Lebanon. Am J Hum Genet 1988; 43: 188–196.
Khlat M : Consanguineous marriages in Beirut: time trends, spatial distribution. Soc Biol 1988; 35: 324–330.
Barbour B, Salameh P : Consanguinity in Lebanon: prevalence, distribution and determinants. J Biosoc Sci 2009; 41: 505–517.
Ten Kate LP, Teeuw M, Henneman L et al: Autosomal recessive disease in children of consanguineous parents: inferences from the proportion of compound heterozygotes. J Community Genet 2010; 1: 37–40.
Gialluisi A, Pippucci T, Anikster Y et al: Estimating the allele frequency of autosomal recessive disorders through mutational records and consanguinity: the Homozygosity Index (HI). Ann Hum Genet 2012; 76: 159–167.
Rosenberg NA, Pemberton TJ, Li JZ et al: Runs of homozygosity and parental relatedness. Genet Med 2013; 15: 753–754.
Rehder CW, David KL, Hirsch B et al: American College of Medical Genetics and Genomics: standards and guidelines for documenting suspected consanguinity as an incidental finding of genomic testing. Genet Med 2013; 15: 150–152.
Acknowledgements
We are grateful to the research council of Saint-Joseph University-Lebanon. This project is supported and funded by the Agence de la Biomedecine (France), Saint-Joseph University (Lebanon) and the French Ministry of Higher Education and Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Jalkh, N., Sahbatou, M., Chouery, E. et al. Genome-wide inbreeding estimation within Lebanese communities using SNP arrays. Eur J Hum Genet 23, 1364–1369 (2015). https://doi.org/10.1038/ejhg.2014.246
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.246
This article is cited by
-
People from Ibiza: an unexpected isolate in the Western Mediterranean
European Journal of Human Genetics (2019)
-
First molecular study in Lebanese patients with Cockayne syndrome and report of a novel mutation in ERCC8 gene
BMC Medical Genetics (2018)
-
Runs of homozygosity: windows into population history and trait architecture
Nature Reviews Genetics (2018)
-
Contribution of copy number variants (CNVs) to congenital, unexplained intellectual and developmental disabilities in Lebanese patients
Molecular Cytogenetics (2015)
