
Abstract
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the gastrointestinal tract, disclosing somatic KIT, PDGFRA and BRAF mutations. Loss of function of succinate dehydrogenase (SDH) complex is an alternative molecular mechanism in GISTs, namely in carriers of germline mutations of the SDH complex that develop Carney–Stratakis dyad characterized by multifocal GISTs and multicentric paragangliomas (PGLs). We studied a series of 25 apparently sporadic primary wild-type (WT) KIT/PDGFRA/BRAF GISTs occurring in patients without personal or familial history of PGLs, re-evaluated clinicopathological features and analyzed molecular alterations and immunohistochemistry expression of SDH complex. As control, we used a series of well characterized 49 KIT/PDGFRA/BRAF-mutated GISTs. SDHB expression was absent in 20% and SDHB germline mutations were detected in 12% of WT GISTs. Germline SDHB mutations were significantly associated to younger age at diagnosis. A significant reduction in SDHB expression in WT GISTs was found when compared with KIT/PDGFRA/BRAF-mutated GISTs. No significant differences were found when comparing DOG-1 and c-KIT expression in WT, SDHB-mutated and KIT/PDGFRA/BRAF-mutated GISTs. Our results confirm the occurrence of germline SDH genes mutations in isolated, apparently sporadic WT GISTs. WT KIT/PDGFRA/BRAF GISTs without SDHB or SDHA/SDHB expression may correspond to Carney–Stratakis dyad or Carney triad. Most importantly, the possibility of PGLs (Carney–Stratakis dyad) and/or pulmonary chondroma (Carney triad) should be addressed in these patients and their kindred.
Similar content being viewed by others


Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the gastrointestinal tract.1 Somatic gain-of-function mutations of v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT) or of platelet-derived growth factor receptor and alpha polypeptide (PDGFRA) genes are the most prevalent molecular alterations occurring in GISTs2, 3, 4 and observed in a mutually exclusive manner.2 KIT mutations are described in 41–92% GISTs5, 6, 7, 8, 9, 10, whereas PDGFRA mutations are present in 5–8% GISTs.11 KIT protein expression is reported in >95% GISTs, including wild-type (WT) tumors and most KIT- and PDGFRA-mutated GISTs.12
Treatment of KIT- and PDGFRA-mutated GISTs with the tyrosine kinase inhibitor (TKI) imatinib mesylate has shown a great efficiency in patients,13 but GISTs without KIT/PDGFRA somatic mutations may be resistant to treatment with imatinib.14, 15 Sunitinib was approved as an alternative TKI for the treatment of patients with resistance or intolerance to imatinib.16 This resistance and/or intolerance to imatinib reinforce the concept that other molecular events rather than KIT or PDGFRA mutations may be implicated in GIST tumorigenesis.
The first studies indicating an alternative pathway involved in GIST tumorigenesis were related to a molecular alteration occurring in the mitogen-activated protein kinase pathway: BRAFV600E somatic mutation was reported in 3–7% KIT/PDGFRA WT GISTs17, 18, 19 and in one KIT-mutated GISTs.20
Recently, loss of function of succinate dehydrogenase (SDH) complex was proposed as another alternative molecular mechanism in KIT/PDGFRA/BRAF WT GISTs.21, 22, 23, 24, 25, 26, 27 In Carney–Stratakis dyad, patients develop multifocal GISTs and paraganglioma (PGL),28, 29 and GISTs are known to display deficient SDH protein expression.22 Further molecular studies addressing Carney–Stratakis syndrome patients disclosed the presence of germline mutations of the SDH subunits B (SDHB), C (SDHC) and D (SDHD).25, 27 These GIST patients do not harbor KIT or PDGFRA mutations.25 In the Carney triad (PGLs, GISTs and pulmonary chondromas)30 patients also present SDH-deficient GISTs22, 23 but SDHA, -B, -C and -D mutations have not been described so far.31, 32
We studied a series of 25 apparently sporadic WT primary KIT/PDGFRA/BRAF GISTs occurring in patients without personal or familial history of PGLs and pulmonary chondromas, re-evaluated their clinicopathological features and evaluated protein expression and molecular alterations of the SDH complex.
Materials and methods
From a series of 78 primary GISTs previously characterized by our group,33 we selected 25 WT KIT/PDGFRA/BRAF GISTs (WT GISTs) with tumors located in the stomach (n=16), small intestine (n=7), colon (n=1) and in undetermined location (n=1). Two WT GISTs (cases 4 and 22) had multifocal presentation, and one (case 22) had metastasis in the colon and omentum. Follow-up of the patients was updated and tumors were re-evaluated according to their clinicopathological features (Table 1). The risk behavior of GIST recurrence was evaluated according to the National Institutes of Health (NIH) risk classification1 and the National Comprehensive Cancer Network (NCCN) risk classification.34
Three patients with WT GISTs were treated with imatinib mesylate. One (case 2) was treated with imatinib mesylate after recurrence of primary tumor, but the patient died due to unrelated comorbidity. Adjuvant imatinib was used in another patient (case 3) that is alive without evidence of recurrence at last follow-up. Neoadjuvant imatinib was used in another patient (case 12) submitted to complete tumor surgical ressection after maximal response, and the patient is alive without evidence of recurrence at last follow-up.
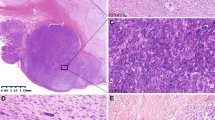
Immunohistochemistry (IHC) for SDHA and SDHB proteins (Figure 1a and b) was performed in representative tumor tissue sections of all 25 WT GISTs, as previously described.21 Negative and positive controls were used simultaneously to ensure specificity and reliability of the staining process. Previously tested positive cases of oncocytic variant of papillary thyroid carcinoma were used as positive controls. Omission of the primary antibody was used as negative control.

SDHA and SDHB protein expression in the adjacent non-tumor gastric mucosa and GIST tumor cells of case 2 (a) and case 4 (b). Note the negative expression of SDHB in the tumor cells of case 2, and the negative expression of SDHB and SDHA in the tumor cells of case 4. Original magnification: × 200. Electropherogram representative of SDHB c.1-10413_73-3866del (c) and SDHB c.T282A mutation (d).
IHC for DOG-1 protein was performed in 23 WT GISTs. Briefly, tissue sections with 2 μm thickness were deparaffinized, rehydrated and pre-treated with 1 × Epitope Retrieval Solution pH 9 (Tris/EDTA-based buffer containing surfactant) (E7119; Leica Microsystems, Newcastle upon Tyne, UK) in a pressure cooker at 98 °C for 1 min, and at 125 °C for 5 min. Immunohistochemical staining was performed with the Novocastra Novolink Polymer Detection System (RE7140-CE; Leica Microsystems), according to the manufacturer’s instructions. The tissue sections were incubated 1 h at room temperature with DOG-1 primary antibody (clone K9, mouse, 1:100 dilution; NCL-L-DOG-1; Leica Microsystems). Detection was performed with Novocastra Novolink Polymer Detection Systems, according to the manufacturer’s instructions, and the samples were developed with DAB chromogen. The slides were mounted using a Richard-Allan Scientific Mounting Medium (Fisher Scientific Gmbh, Schwerte, Germany), after counterstaining with hematoxylin. Expression of DOG-1 was evaluated and compared with the expression of KIT in matched tumor sections. The data regarding KIT expression was reported previously by our group33 in a study comprising the tumors from the present series.
IHC for SDHA, SDHB and DOG-1 protein was performed also in 49 KIT/PDGFRA/BRAF-mutated GISTs from our previously reported series33 that were used as control. IHC evaluation was performed independently by two observers (JML and VM). For SDHA and SDHB protein expression, an IHC score was established, which corresponded to the sum of the intensity of expression (negative=0, faint/moderate=1, strong=2) with the extent of each tumor protein expression (0–5%=0; 5–25%=1, 26–50%=2, 51–75%=3, >75%=4). Expression of SDHA and SDHB was classified as negative, low, moderate and high, when the sum of IHC score was 0, ≤2, 3–4 and >4, respectively.
Tumor DNA was extracted from all 25 WT GISTs as previously described,21 and all the cases were evaluated by PCR and DNA sequencing for the presence of SDHB, SDHC and SDHD point mutations and deletions of exon 1 in SDHB, as previously described.35, 36 Briefly the PCR mixture, 25 μl in total, contained 100 ng of genomic DNA, 1 μl dNTP’s (5 mM each; Bioron GmbH, Ludwigshafen, Germany), 1 μl of each primer (10 μ M),36 5 μl 5 × Green GoTaq Flexi Buffer, 2.5 μl MgCl2 (25 mM) and 0.15 U GoTaq Flexi DNA Polymerase (Promega, Madison, WI, USA). The respective PCR product was purified using the GFX PCR DNA and gel band purification kit (GE Healthcare, Buckinghamshire, UK). The purified PCR product was subjected to automatic sequencing (ABI PRISM 3100 genetic analyzer; Applied Biosystems, Foster City, CA, USA), using the BigDye Terminator version 3.1 cycle sequencing kit (Applied Biosystems). Sense and antisense sequencing was performed. All mutations were further verified by PCR and sequencing from a new DNA template.
In GISTs with mutations of SDHB, SDHC or SDHD, matched adjacent non-tumor tissue DNA was also obtained. Additionally, germline DNA from peripheral blood lymphocytes was obtained from patients in cases 1–5 by the standard proteinase K-SDS digestion and saline precipitation.37 In patients (cases 4 and 5) with SDHA- and SDHB-negative tumors, SDHA mutations were screened in germline DNA of the peripheral blood lymphocytes, as previously described.38 The presence of mutations was validated by a second PCR followed by direct sequencing.
Given that GIST cases 2 and 3 carried SDHB germline mutation, the loss of the other WT allele of the SDHB gene was evaluated. Tumor DNA was analyzed for intragenic deletions using multiplex-ligation-dependent probe amplification (MLPA) assay (SALSA MLPA KIT, P226SDHD, MRC-Holland b.v., Amsterdam, The Netherlands), according to the manufacturer’s instructions. MLPA fragments were discriminated in an ABI PRISM 310 Genetic Analyzer (Applied Biosystems), and the resulting data was analyzed using Coffalyser software (MRC-Holland b.v.). All MLPA results were reproduced at least three times.
Fisher’s exact test, non-parametric Mann–Whitney U test and parametric independent samples t-test were used for the statistical analysis of the results. Specific survival time analysis was determined by Kaplan–Meyer and log-rank tests with SPSS for Windows, V.19.0 (SPSS Inc., Chicago, IL, USA). Values were considered significantly different when P<0.05.
Results
Clinicopathological features of WT GISTs
We studied 25 apparently sporadic primary WT KIT/PDGFRA/BRAF GISTs (WT GISTs) whose clinicopathological features are summarized in Table 1. The gender ratio of the patients (male:female) was 1.8:1. The median age of the patients was 62 (range 26–82) years, the median size of the tumors was 6 cm, and the median mitotic index (mitoses per 50 high-power fields) was 4. The tumors were located in the stomach (n=16), small intestine (n=7), colon (n=1) and undetermined site (n=1). Eight percent (2/25) of the patients had multiple GISTs and one patient (4%–1/25) had metastases in the colon and omentum at presentation. Histologic morphology classification of the tumors revealed epithelioid, spindle and mixed (epithelioid/spindle) GISTs in 20% (5/25), 68% (17/25) and 12% (3/25) of the cases, respectively.
Tumors located in the stomach displayed a significantly higher mean mitotic index (mean 6) than tumors of the small intestine (mean 2) (P=0.022). However, when SDHB-negative WT KIT/PDGFRA/BRAF GISTs (all located in stomach) were excluded from the analysis, no significant difference was detected in the mitotic index between WT KIT/PDGFRA/BRAF GISTs located in the stomach and those located in the small intestine (P=0.094).
The size (19 cm) of the single metastatic tumor was significantly (P=0.04) larger than the mean sizes of the other non-metastatic primary WT GISTs (6.99±0.78 cm) at clinical presentation. Tumors from patients with macroscopic residual tumor (R2; non-curative surgery) revealed significantly (P=0.037) larger mean dimensions (15.75±3.25 cm) than tumors from patients with no macroscopic residual tumor (R0; potentially curative surgery) (6.79±0.89 cm).
Patients with high risk of recurrence according to the NIH classification were significantly (P=0.049) younger (53.0±5.4 years) than patients of lower risk groups (65.6±3.3 years). According to the risk NCCN classification, patients with high risk of recurrence were also younger (52.1±7.5 years) than patients of lower recurrence risk groups (63.8±3.1 years), although not reaching statistical significance (P=0.1).
The mean follow-up of the patients was 114±15 months. At the last follow-up (December, 2011), 15 out of 25 (60%) patients were alive, with no evidence of disease, and 10 patients (40%) were dead. In four patients, deaths were due to GIST progression, and in the remaining by unrelated tumor patient comorbidities. The 5-year specific disease survival of the 25 patients was 83.8%. In the univariate analysis, patients with high-risk tumors (NIH and NCCN classifications) displayed significantly poorer prognosis (P=0.032 and P=0.004, respectively) than patients with lower risk WT GISTs.
It is noteworthy that apart from the aforementioned metastatic lesions, no other tumors (pulmonary chondromas, PGLs) were found in clinical, pathologic and imaging (X-ray and CT) evaluations.
SDHA, SDHB and DOG-1protein expression in GISTs
WT GISTs displayed high (16%), moderate (44%), low (20%) and negative (20%) SDHB expression; and high (68%), moderate (20%), low (4%), and negative (8%) SDHA expression (Figure 2). Five out of the 25 (20%) tumors did not express SDHB (Table 1, Figure 2). Two out of the 5 (40%) SDHB-negative WT GISTs were also negative for SDHA expression.

Frequencies (%) of wild-type and mutated KIT/PDGFRA/BRAF GISTs with negative, low, moderate and high tumor cell expression of SDHA and SDHB proteins.
Patients with SDHB-negative tumors were significantly (P=0.002) younger (41.0±5.9 years) than patients with SDHB-positive tumors (66.0±2.6 years). The mitotic index of GISTs without SDHB expression (mean 9) was significantly (P=0.05) higher than GISTs positive for SDHB expression (mean 4). Absence of SDHB expression was more frequent (P=0.038) in tumors composed exclusively by epithelioid cells.
We evaluated SDHA and SDHB expression in 49 cases of KIT/PDGFRA/BRAF-mutated GISTs previously described by our group,33 and all were positive for both proteins: high (61%), moderate (33%) and low (6%) expression of SDHB; and, high (82%), moderate (16%) and low (2%) expression of SDHA (Figure 2).
The absence of SDHB expression was significantly (P<0.001) associated to WT GISTs when compared with KIT/PDGFRA/BRAF-mutated GISTs. Even excluding WT GISTs negative for SDHB and SDHA/SDHB expression, we found a significant (P=0.003) reduction of SDHB expression in WT GISTs when compared with KIT/PDGFRA/BRAF-mutated GISTs.
In addition, no differences were found in DOG-1 and c-KIT expression when comparing WT, SDHB-mutated and KIT/PDGFRA/BRAF-mutated GISTs. Overall, 93% (69/74) of GISTs expressed KIT and 90% (65/72) expressed DOG-1. In WT GISTs, 92% (23/25) and 83% (19/23) of the tumors expressed KIT and DOG-1, respectively, and 94% (46/49) KIT/PDGFRA/BRAF-mutated GISTs expressed both KIT and DOG-1. Expression of KIT was significantly (P=0.045) associated with DOG-1 expression in this cohort of GIST patients.
SDH molecular alterations in WT GISTs
Four out of 25 (16%) WT GISTs displayed SDHB mutations (Table 1). No somatic or germline SDHC and SDHD mutations were detected in these four cases.
Three patients (12%–3/25) were carriers of germline SDHB mutations (Table 1): two (case 2 and 3) carried a germline deletion encompassing the promoter region and exon 1 of SDHB (c.1-10413_73-3866del; Figure 1c) and one (case 1) carried a germline point mutation in the SDHB (c.T282A; Figure 1d). Case 1 was previously reported by our group.21 None of the germline-mutated cases expressed SDHB in the tumors.
One patient (case 6) presented a tumor with a somatic deletion of promoter and exon 1 of SDHB (c.1-10413_73-3866del). This deletion was absent in the DNA from adjacent non-tumor tissue. SDHB and SDHA expression was detected in the tumor cells of this patient.
Tumor MLPA analysis revealed complete loss of the SDHB promoter and of the exon 1 in the tumor of case 2 (Figure 3). In case 3, MLPA analysis also revealed a complete loss of the SDHB promoter; in addition, we found loss of heterozygosity (LOH) of SDHB in exon 1 in this tumor (Figure 3). In our previous report,21 MLPA analysis indicated LOH of the WT allele in the tumor of case 1.

MLPA quantification of each SDHB exon in DNA extracted from GIST tissue of cases 2 and 3; SDHB promoter and exon 1 display an almost complete loss in the tumor of case 2; in case 3, SDHB promoter displays also an almost complete loss whereas SDHB exon 1 displays LOH in the tumor. Data were normalized using five genomic DNA control samples isolated from normal human tissue. The bars represent the average of three experimental replicas.
SDHA mutations were not found in the germline or tumoral DNA of cases 4 and 5, which did not express SDHA in their GISTs.
Patients with germline mutations in SDHB were significantly (P=0.001) younger (32.7±4.8 years) than patients without germline mutations in SDHB (65.6±2.6 years). The mitotic index of GISTs from patients carrying SDHB germline mutations (mean 12) was significantly (P=0.002) higher than that of patients without germline SDHB mutations (mean 4). However, the presence of germline SDHB mutation was not significantly associated with high risk, according to the NIH (P=0.059) and NCCN (P=0.194) classifications.
Discussion
In the present study, we evaluated the presence of SDH mutations in a series of 25 WT KIT/PDGFRA/BRAF GISTs of patients with no apparent personal or familial history of PGLs and/or pulmonary chondroma. We detected SDHB germline mutations in 12% of the patients. No germline mutations were found in SDHA, SDHC or SDHD. Our results fit with a series recently reported by Janeway et al24 who also identified germline mutations of SDH genes in 12% of seemingly sporadic WT GIST patients.
Twenty percent of WT KIT/PDGFRA/BRAF GISTs did not express SDHB or SDHA/SDHB. Three of the five patients carried germline SDHB mutations. Specifically, one patient (case 1) carried the germline SDHBT282A (p.Ile44Asn) mutation, which was previously reported by our group, and the tumor tissue presented features consistent with LOH of the WT SDHB allele.21 The two remaining patients (cases 2 and 3) carried a germline deletion in the promoter and exon 1 of SDHB (c.1-10413_73-3866del);35 this deletion had previously been identified in PGLs, but this is the first report of its association with the development of GISTs. Case 2 presented complete loss of the promoter and exon 1 of SDHB in the tumor, and case 3 showed complete loss of SDHB promoter and features consistent with LOH of exon 1 of SDHB in the tumor. Altogether, our data fit with the classical two ‘hit’ tumor-suppressor inactivation model in cancer.21, 27, 38
Carney–Stratakis dyad is a familial condition with an apparently autosomal dominant inheritance pattern with incomplete penetrance, characterized by GISTs associated with PGLs.28, 29 In this dyad, germline mutations in SDHB, SDHC or SDHD have been described in the absence of somatic or germline mutations in KIT/PDGFRA.25, 27
The Carney triad is a non-familial condition characterized by GISTs, PGLs and pulmonary chondroma.30, 39 So far, no somatic or germline KIT/PDGFRA mutations, SDHB, SDHC and SDHD mutations, have been described in this triad.31, 32 None of our patients presenting GISTs with loss of SDHB or SDHB/SDHA expression revealed clinical-imaging evidence of other tumors (eg, pulmonary chondromas and/or PGLs).
So far, GISTs from patients with Carney triad and Carney–Stratakis dyad have been reported as negative for SDHB expression.22 In a previous study, we screened for the germline SDHBT282A mutation in case 1 patient family21 and found the same germline mutation in the patient’s mother. As the patient did not show evidence of PGLs, neither his mother of GISTs or PGLs, we hypothesized that this case may represent an incomplete phenotype of the Carney–Stratakis dyad or a rather distinct entity. On the whole, our results raise the issue of whether GIST patients carrying germline mutations in SDHA, -B, -C, or –D are Carney–Stratakis dyad cases with reduced penetrance and/or expressivity of the disease, or if they represent a new hereditary GIST syndrome.
In our series, WT GIST patients with germline mutations in SDHB were significantly younger than WT GIST patients without germline mutations in SDHB. Notably, WT KIT/PDGFRA/BRAF patients with SDHB- or SDHA/SDHB-negative GISTs were also significantly younger than patients with SDHA/SDHB-positive GISTs. Such association between SDH protein complex defects and younger age of patients has been also described in previous reports.22, 23, 24, 26, 40
A significant higher mitotic index was observed in WT KIT/PDGFRA/BRAF GIST negative for SDHB or SDHA/SDHB expression, when compared with WT KIT/PDGFRA/BRAF GISTs with SDHB/SDHA expression. This significant higher mitotic index was even more pronounced in WT KIT/PDGFRA/BRAF GISTs of patient carriers of SDHB germline mutations. Thus, it is possible that loss of SDH complex function may confer a proliferative advantage to GISTs.
In our series, WT KIT/PDGFRA/BRAF GISTs negative for SDHB or SDHA/SDHB expression were all located in the stomach. Such observations has been reported by Gill et al,23 who additionally suggested that negative SDHB GISTs should be classified as type 2. Recently, Doyle et al41 also reported similar observations, although none of the studies performed the molecular characterization of tumors and patients. We observed that WT KIT/PDGFRA/BRAF GISTs located in the stomach showed higher mitotic index than those of the small intestine. However, when WT KIT/PDGFRA/BRAF GISTs with negative expression for SDHB (all located in stomach) were excluded from the analysis, this association was lost suggesting that the mitotic index was associated with SDHB expression. A larger series should be considered to clarify this issue.
The WT GISTs negative for SDHB or SDHA/SDHB displayed epithelioid and spindle cell type morphologies. These findings do not fit with the previously reported exclusive epithelioid or mixed (epithelioid/spindle) cell morphology of GISTs negative for SDHB.22, 23, 40, 41 Our findings however concur with those from Miettinen et al26 and indicate that tumor cells from SDHB-deficient GISTs can display epithelioid, spindle and mixed morphology.
It is noteworthy that we found two WT KIT/PDGFRA/BRAF GISTs with loss of SDHA/SDHB expression and without SDHA, SDHB, SDHC and SDHD mutations, as described in Carney triad.22 However, we did not find evidence of pulmonary chondromas and PGLs in any of these patients. Important, the cases reported in literature with Carney’s triad exhibited variable expression of GISTs, pulmonary chondromas and PGLs.23
Concerning the clinicopathological features of our series, we found a lower gender ratio of the patients with WT GISTs than in previous reports, in which at least half of patients were female.24, 26, 40, 42 Patients with high risk of recurrence of primary WT KIT/PDGFRA/BRAF GISTs, according to the NIH classification,1 were significantly younger than patients of lower recurrence WT GIST risk groups.
All KIT/PDGFRA/BRAF-mutated GISTs used as controls expressed SDHB and SDHA proteins, indicating that molecular alterations in SDH complex do not seem to have a major role in the pathogenesis of KIT/PDGFRA/BRAF-mutated GISTs. Furthermore, when WT-negative SDHB GISTs were excluded from the analysis, WT GISTs displayed decreased SDHB protein expression compared with KIT/PDGFRA/BRAF-mutated GISTs. Our results highlight the role of the SDH complex deregulation as an additional molecular mechanism in WT KIT/PDGFRA/BRAF GISTs,21, 22, 23, 24, 25, 26, 27 albeit diverse processes may drive its deregulation. As far as we are aware there are no reports of concomitant germline SDHx genes mutation in KIT/PDGFRA/BRAF-mutated GISTs, which fits with the positive staining for SDHA/SDHB in KIT/PDGFRA/BRAF-mutated GISTs. However, we cannot rule out the existence of somatic SDHx mutations and further studies should be performed in a series of KIT/PDGFRA/BRAF-mutated GISTs to evaluate (somatic and germline) genetic alterations in SDH complex.
Positive expression of DOG-1 was found in most of the evaluated GISTs, and there were no significant differences in KIT and DOG-1 expression when comparing WT GISTs to KIT/PDGFRA/BRAF-mutated GISTs in our cohort. Expression of KIT in GISTs was significantly associated to DOG-1 expression, suggesting that DOG-1 may be an useful additional marker for the diagnosis of GISTs.43
To summarize, in our series we found that 20% of primary WT KIT/PDGFRA/BRAF GISTs did not express SDH proteins and 12% carried SDHB germline mutations, which were particularly associated with patient’s younger age. The results obtained in our study underline the importance of SDHB and SDHA immunohistochemical screening, particularly in young patients (≤45-year-old) harboring WT KIT/PDGFRA/BRAF GISTs. In negative SDHB and SDHA/SDHB GISTs, patients should be screened for germline mutations of SDHA, -B, -C, and –D genes. Primary GISTs without SDHB or SDHA/SDHB expression may represent a distinctive group that should be considered in the management decisions of these patients (Figure 4). Most importantly, the possibility of coexistent PGLs (Carney—Stratakis dyad) and/or pulmonary chondroma (Carney triad) should be addressed in WT GIST patients and their kindred.

Flowchart for the management of patients with KIT/PDGFRA/BRAF wild-type GISTs.
References
Fletcher CD, Berman JJ, Corless C et al: Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol 2002; 33: 459–465.
Heinrich MC, Corless CL, Duensing A et al: PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003; 299: 708–710.
Hirota S, Isozaki K, Moriyama Y et al: Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998; 279: 577–580.
Lasota J, Miettinen M : KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin Diagn Pathol 2006; 23: 91–102.
Choi YR, Kim H, Kang HJ et al: Overexpression of high mobility group box 1 in gastrointestinal stromal tumors with KIT mutation. Cancer Res 2003; 63: 2188–2193.
Kim NG, Kim JJ, Ahn JY et al: Putative chromosomal deletions on 9P, 9Q and 22Q occur preferentially in malignant gastrointestinal stromal tumors. Int J Cancer 2000; 85: 633–638.
Lasota J, Jasinski M, Sarlomo-Rikala M, Miettinen M : Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol 1999; 154: 53–60.
Lux ML, Rubin BP, Biase TL et al: KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 2000; 156: 791–795.
Rubin BP, Singer S, Tsao C et al: KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001; 61: 8118–8121.
Taniguchi M, Nishida T, Hirota S et al: Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res 1999; 59: 4297–4300.
Rubin BP, Heinrich MC, Corless CL : Gastrointestinal stromal tumour. Lancet 2007; 369: 1731–1741.
Lasota J, Miettinen M : Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology 2008; 53: 245–266.
Demetri GD, von Mehren M, Blanke CD et al: Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002; 347: 472–480.
Heinrich MC, Owzar K, Corless CL et al: Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol 2008; 26: 5360–5367.
Janeway KA, Albritton KH, Van Den Abbeele AD et al: Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr Blood Cancer 2009; 52: 767–771.
Demetri GD, van Oosterom AT, Garrett CR et al: Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 2006; 368: 1329–1338.
Agaimy A, Terracciano LM, Dirnhofer S et al: V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J Clin Pathol 2009; 62: 613–616.
Agaram NP, Wong GC, Guo T et al: Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 2008; 47: 853–859.
Martinho O, Gouveia A, Viana-Pereira M et al: Low frequency of MAP kinase pathway alterations in KIT and PDGFRA wild-type GISTs. Histopathology 2009; 55: 53–62.
Agaimy A, Markl B, Arnholdt H et al: Multiple sporadic gastrointestinal stromal tumours arising at different gastrointestinal sites: pattern of involvement of the muscularis propria as a clue to independent primary GISTs. Virchows Arch 2009; 455: 101–108.
Celestino R, Lima J, Faustino A et al: A novel germline SDHB mutation in a gastrointestinal stromal tumor patient without bona fide features of the Carney-Stratakis dyad. Fam Cancer 2012; 11: 189–194.
Gaal J, Stratakis CA, Carney JA et al: SDHB immunohistochemistry: a useful tool in the diagnosis of Carney-Stratakis and Carney triad gastrointestinal stromal tumors. Mod Pathol 2011; 24: 147–151.
Gill AJ, Chou A, Vilain R et al: Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am J Surg Pathol 2010; 34: 636–644.
Janeway KA, Kim SY, Lodish M et al: Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA 2011; 108: 314–318.
McWhinney SR, Pasini B, Stratakis CA : Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med 2007; 357: 1054–1056.
Miettinen M, Wang ZF, Sarlomo-Rikala M, Osuch C, Rutkowski P, Lasota J : Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol 2011; 35: 1712–1721.
Pasini B, McWhinney SR, Bei T et al: Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 2008; 16: 79–88.
Carney JA, Stratakis CA : Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet 2002; 108: 132–139.
Daum O, Vanecek T, Sima R, Michal M : Gastrointestinal stromal tumor: update. Klinicka Onkologie 2006; 19: 203–211.
Carney JA : Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc 1999; 74: 543–552.
Matyakhina L, Bei TA, McWhinney SR et al: Genetics of carney triad: recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. J Clin Endocrinol Metab 2007; 92: 2938–2943.
Stratakis CA, Carney JA : The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): molecular genetics and clinical implications. J Intern Med 2009; 266: 43–52.
Gomes AL, Gouveia A, Capelinha AF et al: Molecular alterations of KIT and PDGFRA in GISTs: evaluation of a Portuguese series. J Clin Pathol 2008; 61: 203–208.
Miettinen M, Lasota J : Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006; 23: 70–83.
Cascon A, Montero-Conde C, Ruiz-Llorente S et al: Gross SDHB deletions in patients with paraganglioma detected by multiplex PCR: a possible hot spot? Genes Chromosomes Cancer 2006; 45: 213–219.
Lima J, Feijao T, Ferreira da Silva A et al: High frequency of germline succinate dehydrogenase mutations in sporadic cervical paragangliomas in northern Spain: mitochondrial succinate dehydrogenase structure-function relationships and clinical-pathological correlations. J Clin Endocrinol Metab 2007; 92: 4853–4864.
Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Burnichon N, Briere JJ, Libe R et al: SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet 2010; 19: 3011–3020.
Carney JA, Sheps SG, Go VL, Gordon H : The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. N Engl J Med 1977; 296: 1517–1518.
Pantaleo MA, Nannini M, Astolfi A, Biasco G : A distinct pediatric-type gastrointestinal stromal tumor in adults: potential role of succinate dehydrogenase subunit A mutations. Am J Surg Pathol 2011; 35: 1750–1752.
Doyle LA, Nelson D, Heinrich MC, Corless CL, Hornick JL : Loss of sdhb expression is limited to a distinctive subset of gastric wild-type gastrointestinal stromal tumors: a comprehensive genotype-phenotype correlation study. Histopathology 2012, no-no.
Rege TA, Wagner AJ, Corless CL, Heinrich MC, Hornick JL : ‘Pediatric-type’ gastrointestinal stromal tumors in adults: distinctive histology predicts genotype and clinical behavior. Am J Surg Pathol 2011; 35: 495–504.
West RB, Corless CL, Chen X et al: The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol 2004; 165: 107–113.
Acknowledgements
This study was supported by Fundação Calouste Gulbenkian through a PhD grant to RC; by Fundação para a Ciência e Tecnologia through the program Ciência 2007 (VM) and 2008 (JL) and partially by Novartis Oncology, Portugal, through a project grant. IPATIMUP is an Associate Laboratory of the Portuguese Ministry of Science, Technology and Higher Education that is partially supported by the FCT. We thank to Menarini Diagnósticos for kindly providing the DOG-1 antibody. We are grateful to Joana Pinto Couto Silva for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Celestino, R., Lima, J., Faustino, A. et al. Molecular alterations and expression of succinate dehydrogenase complex in wild-type KIT/PDGFRA/BRAF gastrointestinal stromal tumors. Eur J Hum Genet 21, 503–510 (2013). https://doi.org/10.1038/ejhg.2012.205
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.205
Keywords
This article is cited by
-
Wild-Type Gastrointestinal Stromal Tumors—Molecular Features, Frequency, and Consequences Among the Indian Population
Indian Journal of Surgery (2024)
-
Low frequency of TERT promoter mutations in gastrointestinal stromal tumors (GISTs)
European Journal of Human Genetics (2015)
-
ETV1 mRNA is specifically expressed in gastrointestinal stromal tumors
Virchows Archiv (2015)
-
Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST
European Journal of Human Genetics (2014)
-
Frequency of TERT promoter mutations in human cancers
Nature Communications (2013)
