
Abstract
Migraine is a common neurological disorder with a genetically complex background. This paper describes a meta-analysis of genome-wide association (GWA) studies on migraine, performed by the Dutch–Icelandic migraine genetics (DICE) consortium, which brings together six population-based European migraine cohorts with a total sample size of 10 980 individuals (2446 cases and 8534 controls). A total of 32 SNPs showed marginal evidence for association at a P-value<10−5. The best result was obtained for SNP rs9908234, which had a P-value of 8.00 × 10−8. This top SNP is located in the nerve growth factor receptor (NGFR) gene. However, this SNP did not replicate in three cohorts from the Netherlands and Australia. Of the other 31 SNPs, 18 SNPs were tested in two replication cohorts, but none replicated. In addition, we explored previously identified candidate genes in the meta-analysis data set. This revealed a modest gene-based significant association between migraine and the metadherin (MTDH) gene, previously identified in the first clinic-based GWA study (GWAS) for migraine (Bonferroni-corrected gene-based P-value=0.026). This finding is consistent with the involvement of the glutamate pathway in migraine. Additional research is necessary to further confirm the involvement of glutamate.
Similar content being viewed by others
Introduction
Migraine is a common neurological disorder that is characterized by severe attacks of headache accompanied by symptoms such as nausea, vomiting and photophobia and phonophobia.1 Two main types of migraine are distinguished based on the presence of an aura that can precede the headache: migraine with aura (MA) or without aura (MO). Although MA and MO have been considered distinct disease entities,2, 3 it is now more and more accepted that they represent different manifestations of the same disease.4, 5, 6
Genetic studies in familial hemiplegic migraine (FHM), a rare monogenic subtype of MA that is considered a suitable model for common migraine,7 revealed three genes (CACNA1A,8 ATP1A29 and SCN1A10) that are involved in ion and neurotransmitter transport in the brain. Despite considerable efforts, linkage and candidate-gene association studies in common migraine have had limited success, with only a few consistently replicated linkage findings.11, 12, 13, 14, 15, 16 A recent genome-wide association study (GWAS), using data from migraine patients who were recruited through headache clinics, found evidence for a role of the metadherin (MTDH) gene in common migraine.17 The associated SNP in that study affects MTDH gene expression and thereby indirectly regulates the expression of the glutamate transporter gene SLC1A2 (also known as EAAT2 or GLT-1), encoding a major glutamate transporter in the brain. This fits in well with the theory that increased glutamate release or reduced glutamate uptake increases the risk of migraine attacks.18, 19, 20, 21, 22
Here, we present a GWA meta-analysis for common migraine by the Dutch Icelandic migraine genetics consortium (DICE). This is the first population-based GWAS for common migraine, including 2446 migraine cases and 8534 controls from six Dutch and Icelandic samples. For replication, two population-based samples of Dutch and one of Australian origin were available. De novo genotyping was performed in the two Dutch replication cohorts (N=769 and 337 cases; 940 and 826 controls, respectively). In addition, an in silico replication study was performed in the Australian replication cohort (N=1851 cases, 4008 controls).
Methods
Populations: subjects, phenotypes and genotyping
The five Dutch samples that were used for the meta-analysis came from the Erasmus Rucphen Family (ERF) study,23, 24 The Netherlands Study of Depression and Anxiety (NESDA),25 The Netherlands Twin Registry (‘NTR1’ and ‘NTR2’)26 and the Rotterdam study,27 and included 330, 756, 378, 276 and 349 migraine cases, respectively. The Icelandic sample came from the AGES-Reykjavik Study and included 357 migraine cases.28 In addition to the 2446 migraine cases, 8534 non-migraine controls (2862 Icelandic and 5672 Dutch controls) from the respective cohorts were included (for details, see Table 1 ). All individuals came from population-based samples and were unrelated, with the exception of the ERF participants, who are part of a genetically isolated population in the Southwest of the Netherlands. Data on migraine symptomatology were collected by means of questionnaires (ie, AGES, NESDA, NTR1–2, Rotterdam), or a combination of questionnaires and telephone interview follow-up (ie, ERF).
Three additional independent samples were available for replication; two Dutch population-based samples (the GEM sample29 and a third sample from the NTR), and one Australian sample, the Australian Twin Migraine (ATM) GWA study.30, 31 The Dutch GEM sample included 769 migraine cases and 940 non-migraine controls. The NTR replication sample consisted of 337 cases and 826 non-migraine controls, and the Australian sample consisted of 5859 unrelated individuals (1851 migraine cases, 1631 non-migraine controls and 2377 additional unselected controls).
Genotyping was performed using a variety of SNP genotyping platforms. To ensure sufficient overlap between studies?, genotypes for ∼2.5 million HapMap SNPs were imputed using MACH32 or IMPUTE33 software. An overview of the samples, including details on sample size, genotyping and imputations, is provided in Table 1. More details on the background of the studies, phenotyping strategies and genotyping procedures can be found in the Supplementary data.
GWA and meta-analysis
In each sample, a logistic regression association test was carried out. Next, a meta-analysis was performed combining the GWA results of the six samples (total number of individuals: 10 890) using the METAL program (http://www.sph.umich.edu/csg/abecasis/metal/). As different phenotype definitions were used in the different samples, the effect sizes may not be directly comparable between studies. Therefore, a pooled Z-score approach was used. With the pooled Z-score method, an overall Z-score is calculated based on the summed Z-scores from the individual studies, weighted by each study's sample size. The weights are calculated as the square root of (Nstudy/Ntotal), and the squared weights sum to one. The direction of effect is indicated by the sign of the Z-score. To ensure that meta-analysis results were based on SNP data of a large enough number of individuals, 184 350 SNPs that were available for <70% of all participants were excluded from the meta-analysis. This left a total of 2 394 913 autosomal SNPs for analysis. Annotation of meta-analysis results was performed with WGA viewer version 1.26E (Dongliang Ge and David B Goldstein; http://people.genome.duke.edu/~dg48/WGAViewer/).34 P-values<5 × 10−8 were considered genome-wide significant.35
Replication studies
A replication study was performed with direct genotyping in the GEM and the NTR replication samples. The top SNP was genotyped with a TaqMan assay in both GEM and the NTR replication sample. In addition, another 18 SNPs with a P-value<1 × 10−5 were selected based on informativeness given the LD structure. These SNPs were genotyped in the GEM sample using an in-house Sequenom iPLEX Mass-ARRAY platform (Sequenom Inc., San Diego, CA, USA). Logistic regression was performed to test for association between these SNPs and migraine status. Third, all DICE SNPs with a P-value<1 × 10−4 in the meta-analysis were selected, and for these SNPs, an in silico replication study was performed in the ATM GWA data set. Finally, the 19 SNPs that were genotyped in GEM and the NTR replication sample and measured or imputed in the ATM GWA replication sample were meta-analysed together with the discovery data sets. A more detailed description of the genotyping procedures and association analyses can be found in the Supplementary data.
Post hoc analyses
Text mining
Relationships between genes (emerging from the meta-analysis) and migraine were studied using the Anni text-mining program (Anni version 2.1; http://www.biosemantics.org/anni).36 For details see the Supplementary data.
Comparison of results with migraine genes and loci from previous studies
Genome-wide linkage studies and association studies for migraine were identified with a literature search in PubMed. We examined which SNPs with a P-value<1 × 10−4 coincided with a region containing a published migraine linkage peak. In addition, a selection of migraine candidate genes was made and inspected in our meta-analysis data set by calculating a gene-based P-value for each of the selected genes using the VEGAS program.37 More details can be found in the Supplementary methods.
Results
Meta-analysis
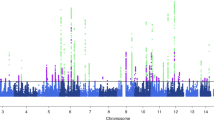
GWA analyses were performed in the six population-based samples and the results were meta-analyzed using a pooled Z-score approach. As shown in Figure 1, no systematic deviation from the expected distribution of P-values was observed in the Q–Q plot, which is reflected by a genomic inflation factor (λ) of 1.022. A total of 32 SNPs had a P-value<1 × 10−5 (Table 2 ). None of these SNPs exceeded the threshold for genome-wide significance (Figure 2). A total of 10 SNPs were located within genes; 8 in the metastasis associated in colon cancer 1 (MACC1) gene (7p21), 1 in the immunoglobulin lambda-like polypeptide 1 (IGLL1) gene (22q11) and 1 in the nerve growth factor receptor (NGFR) gene (17q21–q22). The most significant result was obtained for SNP rs9908234 (P=8.00 × 10−8) in the NGFR gene, with the strongest evidence coming from the AGES and NESDA studies (Supplementary Table S1). Data for 17 additional SNPs in this gene were available, but none of these were associated with migraine (all P-values>0.05). These SNPs were not in LD with rs9908234. Next, we performed text mining with the Anni program. The concept ‘migraine’ was matched against the genes located within or close to our top SNPs (P-value<1 × 10−4). Remarkably, the NGFR gene surfaced as the best migraine candidate gene.

Q–Q plot showing the expected and observed distribution of P-values in the meta-analysis that included the five Dutch samples and the Icelandic sample. The genomic inflation factor (λ) for the meta-analysis was 1.022.

Manhattan plot showing the P-values by chromosome for the meta-analysis.
Replication analysis of the top SNPs
From the 32 top SNPs with P-values<1 × 10−5, we selected 19 SNPs for genotyping in the GEM sample. The selection was made such that the genotyped SNPs were maximally informative given the LD between them. The top SNP rs9908234, located in the NGFR gene, was genotyped in one additional replication sample from the NTR. The association observed in the discovery samples could not be replicated for rs9908234 (GEM: OR=0.86, P=0.31; NTR replication sample: OR=0.89, P=0.579; see Supplementary Table S2). The findings for the other 18 SNPs were not replicated either. None showed a P-value<0.05 in the GEM sample: the smallest P-value observed was 0.10, but this effect was in the opposite direction compared with the meta-analysis.
An in silico replication study was performed in the ATM GWA sample. This analysis included all SNPs with P-values <1 × 10−4 in the DICE meta-analysis. In the ATM GWA sample, there were data for 327 out of 340 SNPs with P-values<1 × 10−4. None of these SNPs had a P-value<0.01 in the ATM GWA data set. A total of 11 SNPs had P-values between 0.01 and 0.05, but for only 3 SNPs, the effect was in the same direction as in the DICE cohorts (rs6919479, P=0.045; rs9363693, P=0.045; and rs9294736, P=0.037; all on chromosome 6). These results were not significant after correction for multiple testing.
Finally, a new meta-analysis was carried out in the DICE discovery samples, the GEM sample and the ATM GWA sample, for the 19 SNPs genotyped in GEM. For rs9908234, the meta-analysis also included the NTR replication cohort. The P-values for these SNPs did not decrease compared with the first meta-analysis (Supplementary Table S2).
Comparison of meta-analysis results with previous genetic findings in migraine
The large sample size of the present study provided a unique opportunity to further investigate previous findings from linkage and candidate gene studies on a larger scale, and to try and replicate the findings recently reported in a large clinic-based GWAS for migraine.17
First, we investigated whether there were any SNPs with P-values<10−4 that were located in previously identified migraine linkage regions (Supplementary Table S3). Five SNPs were located on chromosome 10q22–q23, a locus that has been reported for migraine several times.11, 15, 16 However, none were located in or near a gene that could easily be linked to migraine pathophysiology. Interestingly, one SNP (rs1972860, P=6.02 × 10−5) was located in the glutamate receptor, ionotropic, delta 2 (GRID2) gene on chromosome 4q22, a region reported in several different migraine linkage studies.11, 12, 13, 14
In addition, we performed a gene-based association test for selected candidate genes for migraine (Table 3 ). Seven candidate genes were selected based on the results of previous candidate gene association studies for common migraine. Furthermore, a recently published GWAS of clinic-based migraine identified an SNP (rs1835740) that was located between two interesting candidate genes: the MTHD gene and the plasma glutamate carboxypeptidase (PGCP) gene.17 An eQTL analysis revealed that rs1835740 most likely affects migraine through cis-regulation of MTHD, which in turn downregulates SLC1A2, a gene that encodes an important glutamate transporter in the brain. Therefore, we selected MTHD, PGCP and SLC1A2 as candidate genes, and also inspected SNP rs1835740 and two nearby correlated SNPs (rs982502 and rs2436046). Finally, the three FHM genes (CACNA1A, ATP1A2 and SCN1A) were included in the analysis.
Gene-based tests were performed for each of the selected candidate genes, using the meta-analysis results of all SNPs tested in the respective genes (Table 3). A gene-based test result was considered significant at an alpha level of 0.05, with Bonferroni correction for 13 tests, which corresponds to a gene-based P-value of 0.05/13=0.0038. None of the genes identified through candidate gene association studies was significantly associated with migraine in the meta-analysis. Although there were nominally significant SNPs in the LTA, ESR1 and INSR genes, results were not significant after correction for the number of SNPs tested within the respective genes. The PGCP and SLC1A2 genes also had several nominally significant SNPs, but again were not significant in the gene-based test. However, in the MTHD gene, 19 of the 28 tested SNPs had a P-value<0.01 in the meta-analysis (Supplementary Table S4). The gene-based P-value for MTDH was 0.002, which remained significant after Bonferroni correction. The SNP that showed association in the clinic-based GWA study (rs1835740)17 did not show significant association with migraine in the meta-analysis (P=0.64). Two nearby SNPs (rs982502 and rs2436046) reported in the same GWAS were also not associated with migraine in the present study.
Finally, we tested the three FHM genes, and found several nominally significant SNPs within CACNA1A and ATP1A2. The gene-based test for CACNA1A (best SNP rs3764615, P=0.004) was not significant (P=0.30). The gene-based P-value for ATP1A2 (best SNP rs2854248, P=3.62 × 10−4) was 0.006.
Discussion
This study describes the first meta-analysis of GWAS for population-based migraine, and contains a total of 2446 migraine cases and 8534 controls. The best P-value was obtained for SNP rs9908234, which is located in the NGFR gene. A replication study was performed in two Dutch replication cohorts that were available for wet replication; the GEM cohort (769 cases, 940 controls) and the NTR replication cohort (337 cases, 826 controls). In addition, the ATM GWA cohort (1851 cases, 1631 controls) was available for in silico replication. Although the NGFR gene is an interesting candidate gene for migraine, the association of NGFR with migraine could not be replicated in these cohorts. A total of 18 additional top SNPs (P-value<10−5) from the meta-analysis were tested in the GEM cohort and the ATM GWA cohort, but none could be replicated successfully.
There are several possible explanations for the lack of replication. First, several different genotyping platforms were used, which made imputation necessary to ensure sufficient overlap between the studies. Also, two different programs (MACH and IMPUTE) were used for imputation. However, given that MACH and IMPUTE use very similar imputation algorithms, and have been reported to be very similar in imputation accuracy,38 we do not expect this to have a major effect on our results. Second, there were some differences between the samples in the precision of the migraine diagnoses, and in most samples, a clinical migraine diagnosis was not available. This is often the case in population-based studies because, for reasons of efficiency, diagnoses are commonly made with (short) headache questionnaires. Unlike in clinic-based studies, they are not usually further evaluated with more extensive questionnaires or interviews by specialized physicians. Less accuracy of diagnosis may result in reduced power to detect association. The phenotypic differences also extend to the control groups, as all non-migraine individuals were included as controls. These differences between studies mean that effect sizes may not be directly comparable. To address this, a pooled Z-score meta-analysis was performed. This type of analysis does not require a direct comparison of effect sizes.39 Third, population-based cohorts also include many patients who have less severe migraine and a lower attack frequency. This means that they might be a genetically more heterogeneous group than patients from clinic-based cohorts. In addition, they are likely to have a lower genetic risk of migraine than the more severely affected patients in clinic-based cohorts. As a consequence, population-based studies may require a larger number of patients for sufficient power. Given that this study replicates previous findings, but does not produce genome-wide significant results, insufficient power (possibly because of the reasons above) seems the most likely explanation for the lack of replication of our top results. A lack of power makes it difficult to distinguish between true associations and false-positive findings in the original meta-analysis. Therefore, when the discovery samples have insufficient power, SNPs selected for replication based on small P-values may not replicate (even in sufficiently large replication samples) because they are false positives. Finally, it should be mentioned that the NESDA sample differed from the other samples because the majority of NESDA participants were selected for major depression. Because of the comorbidity of migraine and major depression, there is a higher prevalence of migraine in this sample than in the other samples. However, given that the percentage of MDD was similar in the migraine cases and the controls (94.3 vs 86.6%), any associations detected in this sample will be related to migraine and not to MDD.
In the present study, we also investigated SNP rs1835740 that was found to be significantly associated with MA in the first GWA study of clinic-based populations.17 This SNP is located on 8q21 between the MTDH and PGCP genes. The SNP itself was not associated with migraine in our study, but our gene-based analyses provided modest support for an association of MTDH with migraine.
In summary, although this study does not provide genome-wide significant association of an SNP with migraine, it provides suggestive evidence for an association with the MTDH gene, which is involved in the glutamate pathway, previously hypothesized to have a role in migraine based on findings in FHM.40 Clearly, even though a large number of patients and controls were included, the present study suffered from a lack of power. In addition to simply increasing the sample size, additional strategies aimed at minimizing phenotypic and genetic heterogeneity may be necessary. Strategies to achieve this can include the identification of reliable biomarkers or stratification of samples based on phenotypic similarity (eg, by looking at trait components,11 specific symptoms16, 41 and/or comorbid pathology). In addition, in future studies it may be worth focusing specifically on the glutamate pathway to assess whether genetic variants affecting glutamate levels are systematically associated with migraine.
References
Headache classification subcommittee of the International Headache Society: The International Classification of Headache Disorders, 2nd edn. Cephalalgia, 2004, Vol 24 (Suppl 1), pp 9–160.
Russell MB, Rasmussen BK, Fenger K, Olesen J : Migraine without aura and migraine with aura are distinct clinical entities: a study of four hundred and eighty-four male and female migraineurs from the general population. Cephalalgia 1996; 16: 239–245.
Russell MB, Ulrich V, Gervil M, Olesen J : Migraine without aura and migraine with aura are distinct disorders. A population-based twin survey. Headache 2002; 42: 332–336.
Kallela M, Wessman M, Havanka H, Palotie A, Farkkila M : Familial migraine with and without aura: clinical characteristics and co-occurrence. Eur J Neurol 2001; 8: 441–449.
Ligthart L, Boomsma DI, Martin NG, Stubbe JH, Nyholt DR : Migraine with aura and migraine without aura are not distinct entities: further evidence from a large Dutch population study. Twin Res Hum Genet 2006; 9: 54–63.
Nyholt DR, Gillespie NG, Heath AC, Merikangas KR, Duffy DL, Martin NG : Latent class and genetic analysis does not support migraine with aura and migraine without aura as separate entities. Genet Epidemiol 2004; 26: 231–244.
Ferrari MD, Van den Maagdenberg AM, Frants RR, Goadsby PJ : Migraine as a cerebral ionopathy with impaired central sensory processing; in Waxman SG (ed): Molecular Neurology. Amsterdam: Elsevier, 2007, pp 439–461.
Ophoff RA, Terwindt GM, Vergouwe MN et al: Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996; 87: 543–552.
De Fusco M, Marconi R, Silvestri L et al: Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet 2003; 33: 192–196.
Dichgans M, Freilinger T, Eckstein G et al: Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 2005; 366: 371–377.
Anttila V, Kallela M, Oswell G et al: Trait components provide tools to dissect the genetic susceptibility of migraine. Am J Hum Genet 2006; 79: 85–99.
Oedegaard KJ, Greenwood TA, Lunde A, Fasmer OB, Akiskal HS, Kelsoe JR : A genome-wide linkage study of bipolar disorder and co-morbid migraine: replication of migraine linkage on chromosome 4q24, and suggestion of an overlapping susceptibility region for both disorders on chromosome 20p11. J Affect Disord 2010; 122: 14–26.
Wessman M, Kallela M, Kaunisto MA et al: A susceptibility locus for migraine with aura, on chromosome 4q24. Am J Hum Genet 2002; 70: 652–662.
Bjornsson A, Gudmundsson G, Gudfinnsson E et al: Localization of a gene for migraine without aura to chromosome 4q21. Am J Hum Genet 2003; 73: 986–993.
Anttila V, Nyholt DR, Kallela M et al: Consistently replicating locus linked to migraine on 10q22-q23. Am J Hum Genet 2008; 82: 1051–1063.
Nyholt DR, Morley KI, Ferreira MA et al: Genomewide significant linkage to migrainous headache on chromosome 5q21. Am J Hum Genet 2005; 77: 500–512.
Anttila V, Stefansson H, Kallela M et al: Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat Genet 2010; 42: 869–873.
Andreou AP, Goadsby PJ : Therapeutic potential of novel glutamate receptor antagonists in migraine. Expert Opin Investig Drugs 2009; 18: 789–803.
Ferrari MD : Migraine. Lancet 1998; 351: 1043–1051.
Goadsby PJ, Classey JD : Glutamatergic transmission in the trigeminal nucleus assessed with local blood flow. Brain Res 2000; 875: 119–124.
van den Maagdenberg AM, Haan J, Terwindt GM, Ferrari MD : Migraine: gene mutations and functional consequences. Curr Opin Neurol 2007; 20: 299–305.
Martinez F, Castillo J, Rodriguez JR, Leira R, Noya M : Neuroexcitatory amino acid levels in plasma and cerebrospinal fluid during migraine attacks. Cephalalgia 1993; 13: 89–93.
Sleegers K, de Koning I, Aulchenko YS et al: Cerebrovascular risk factors do not contribute to genetic variance of cognitive function: the ERF study. Neurobiol Aging 2007; 28: 735–741.
Stam AH, de Vries B, Janssens AC et al: Shared genetic factors in migraine and depression. Evidence from a genetic isolate. Neurology 2010; 74: 288–294.
Penninx BW, Beekman AT, Smit JH et al: The Netherlands Study of Depression and Anxiety (NESDA): rationale, objectives and methods. Int J Methods Psychiatr Res 2008; 17: 121–140.
Boomsma DI, de Geus EJ, Vink JM et al: Netherlands Twin Register: from twins to twin families. Twin Res Hum Genet 2006; 9: 849–857.
Hofman A, Breteler MM, van Duijn CM et al: The Rotterdam Study: objectives and design update. Eur J Epidemiol 2007; 22: 819–829.
Scher AI, Gudmundsson LS, Sigurdsson S et al: Migraine headache in middle age and late-life brain infarcts. JAMA 2009; 301: 2563–2570.
Launer LJ, Terwindt GM, Ferrari MD : The prevalence and characteristics of migraine in a population-based cohort: the GEM study. Neurology 1999; 53: 537–542.
Medland SE, Zayats T, Glaser B et al: A variant in LIN28B is associated with 2D:4D finger-length ratio, a putative retrospective biomarker of prenatal testosterone exposure. Am J Hum Genet 2010; 86: 519–525.
Nyholt DR, LaForge KS, Kallela M et al: A high-density association screen of 155 ion transport genes for involvement with common migraine. Hum Mol Genet 2008; 17: 3318–3331.
Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR : MaCH: Using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 2006; 34: 816–834.
Marchini J, Howie B, Myers S, McVean G, Donnelly P : A new multipoint method for genome-wide association studies by imputation of genotypes. Nature genetics 2007; 39: 906–913.
Ge D, Zhang D, Need AC et al: WGAViewer: Software for Genomic Annotation of Whole Genome Association Studies. Genome Res 2008; 18: 640–643.
Dudbridge F, Gusnanto A : Estimation of significance thresholds for genomewide association scans. Genet Epidemiol 2008; 32: 227–234.
van Haagen HH, t Hoen PA, Botelho Bovo A et al: Novel protein-protein interactions inferred from literature context. PLoS One 2009; 4: e7894.
Liu JZ, McRae AF, Nyholt DR et al: A versatile gene-based test for genome-wide association studies. Am J Hum Genet 2010; 87: 139–145.
Nothnagel M, Ellinghaus D, Schreiber S, Krawczak M, Franke A : A comprehensive evaluation of SNP genotype imputation. Hum Genet 2009; 125: 163–171.
De Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF : Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet 2008; 17: R122–R128.
De Vries B, Frants RR, Ferrari MD, van den Maagdenberg AM : Molecular genetics of migraine. Hum Genet 2009; 126: 115–132.
Ligthart L, Nyholt DR, Hottenga JJ, Distel MA, Willemsen G, Boomsma DI : A genome-wide linkage scan provides evidence for both new and previously reported loci influencing common migraine. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 1186–1195.
Schurks M, Rist PM, Kurth T : MTHFR 677C>T and ACE D/I polymorphisms in migraine: a systematic review and meta-analysis. Headache 2009; 50: 588–599.
Schurks M, Zee RY, Buring JE, Kurth T : Interrelationships among the MTHFR 677C>T polymorphism, migraine, and cardiovascular disease. Neurology 2008; 71: 505–513.
Schurks M, Zee RY, Buring JE, Kurth T : MTHFR 677C → T and ACE D/I polymorphisms and migraine attack frequency in women. Cephalalgia 2010; 30: 447–456.
Rubino E, Ferrero M, Rainero I, Binello E, Vaula G, Pinessi L : Association of the C677T polymorphism in the MTHFR gene with migraine: a meta-analysis. Cephalalgia 2009; 29: 818–825.
Scher AI, Terwindt GM, Verschuren WM et al: Migraine and MTHFR C677T genotype in a population-based sample. Ann Neurol 2006; 59: 372–375.
Oterino A, Valle N, Pascual J et al: Thymidylate synthase promoter tandem repeat and MTHFD1 R653Q polymorphisms modulate the risk for migraine conferred by the MTHFR T677 allele. Brain Res 2005; 139: 163–168.
Lee KA, Jang SY, Sohn KM et al: Association between a polymorphism in the lymphotoxin-a promoter region and migraine. Headache 2007; 47: 1056–1062.
Asuni C, Stochino ME, Cherchi A et al: Migraine and tumour necrosis factor gene polymorphism. An association study in a Sardinian sample. J Neurol 2009; 256: 194–197.
Rainero I, Grimaldi LM, Salani G et al: Association between the tumor necrosis factor-alpha -308 G/A gene polymorphism and migraine. Neurology 2004; 62: 141–143.
Oterino A, Toriello M, Cayon A et al: Multilocus analyses reveal involvement of the ESR1, ESR2, and FSHR genes in migraine. Headache 2008; 48: 1438–1450.
Oterino A, Pascual J, Ruiz de Alegria C et al: Association of migraine and ESR1 G325C polymorphism. Neuroreport 2006; 17: 61–64.
Kaunisto MA, Kallela M, Hamalainen E et al: Testing of variants of the MTHFR and ESR1 genes in 1798 Finnish individuals fails to confirm the association with migraine with aura. Cephalalgia 2006; 26: 1462–1472.
Lea RA, Dohy A, Jordan K, Quinlan S, Brimage PJ, Griffiths LR : Evidence for allelic association of the dopamine beta-hydroxylase gene (DBH) with susceptibility to typical migraine. Neurogenetics 2000; 3: 35–40.
Fernandez F, Colson N, Quinlan S, MacMillan J, Lea RA, Griffiths LR : Association between migraine and a functional polymorphism at the dopamine beta-hydroxylase locus. Neurogenetics 2009; 10: 199–208.
Fernandez F, Lea RA, Colson NJ, Bellis C, Quinlan S, Griffiths LR : Association between a 19 bp deletion polymorphism at the dopamine beta-hydroxylase (DBH) locus and migraine with aura. J Neurol Sci 2006; 251: 118–123.
Paterna S, Di Pasquale P, D’Angelo A et al: Angiotensin-converting enzyme gene deletion polymorphism determines an increase in frequency of migraine attacks in patients suffering from migraine without aura. Eur Neurol 2000; 43: 133–136.
McCarthy LC, Hosford DA, Riley JH et al: Single-nucleotide polymorphism alleles in the insulin receptor gene are associated with typical migraine. Genomics 2001; 78: 135–149.
Acknowledgements
The Age, Gene/Environment Susceptibility Reykjavik Study is funded by NIH contract N01-AG-12100, the NIA Intramural Research Program, Hjartavernd (the Icelandic Heart Association) and the Althingi (the Icelandic Parliament). The Erasmus Rucphen Family was supported by grants from The Netherlands Organization for Scientific Research (NWO), Erasmus MC and the Netherlands Genomics Initiative (NGI)-sponsored Center of Medical Systems Biology (CMSB). The genotyping for the ERF study was supported by EUROSPAN (European Special Populations Research Network) through the European Commission FP6 STRP grant (018947; LSHG-CT-2006-01947). The ERF study was further supported by grants from the Netherlands Organisation for Scientific Research (NWO) (903-52-291, M.D.F., Vici 918.56.602, M.D.F, 907-00-217 G.M.T.), Erasmus MC, the Centre for Medical Systems Biology (CMSB1 and CMSB2), and the Netherlands Genomics Initiative (NGI). We are grateful to all patients and their relatives, general practitioners and neurologists for their contributions and to P Veraart for her help in genealogy, Jeannette Vergeer for the supervision of the laboratory work and P Snijders for his help in data collection. For NESDA and NTR, funding was obtained from the Netherlands Organization for Scientific Research (NWO: MagW/ZonMW): Genetic basis of anxiety and depression (904-61-090); Genetics of individual differences in smoking initiation and persistence (NWO 985-10-002); Resolving cause and effect in the association between exercise and well-being (904-61-193); Twin family database for behavior genomics studies (480-04-004); Twin research focusing on behavior (400-05-717); Genetic determinants of risk behavior in relation to alcohol use and alcohol use disorder (Addiction-31160008); Genotype/phenotype database for behavior genetic and genetic epidemiological studies (40-0056-98-9032); Spinozapremie (SPI 56-464-14192); CMSB: Center for Medical Systems Biology (NWO Genomics); NBIC/BioAssist/RK/2008.024); BBMRI –NL: Biobanking and Biomolecular Resources Research Infrastructure (184.021.007); the VU University: Institute for Health and Care Research (EMGO+ ) and Neuroscience Campus Amsterdam (NCA); the European Science Foundation (ESF): Genomewide analyses of European twin and population cohorts (EU/QLRT-2001-01254); European Community's Seventh Framework Program (FP7/2007-2013): ENGAGE (HEALTH-F4-2007-201413); the European Science Council (ERC) Genetics of Mental Illness (230374); Rutgers University Cell and DNA Repository cooperative agreement (NIMH U24 MH068457-06); Collaborative study of the genetics of DZ twinning (NIH R01D0042157-01A); the Genetic Association Information Network, a public–private partnership between the NIH and Pfizer Inc., Affymetrix Inc. and Abbott Laboratories. The infrastructure for the NESDA study (http://www.nesda.nl) is funded through the Geestkracht program of the Netherlands Organization for Health Research and Development (ZonMw, Grant number 10-000-1002) and is supported by participating universities and mental health care organizations (VU University Medical Center, GGZ inGeest, Arkin, Leiden University Medical Center, GGZ Rivierduinen, University Medical Center Groningen, Lentis, GGZ Friesland, GGZ Drenthe, Scientific Institute for Quality of Health Care (IQ Healthcare), Netherlands Institute for Health Services Research (NIVEL) and Netherlands Institute of Mental Health and Addiction (Trimbos)). SNP genotyping was funded by the Genetic Association Information Network (GAIN) of the Foundation for the US National Institutes of Health (NTR1/NESDA) and the Spinozapremie (NWO/SPI 56-464-14192; NTR2). Statistical analyses were partly carried out on the Genetic Cluster Computer (NWO 480-05-003). The Rotterdam Study (I, I, and II) are funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), the Netherlands Genomics Initiative (NGI)-sponsored Netherlands Consortium for Healthy Aging (NCHA) and the Municipality of Rotterdam. The generation and management of GWAS genotype data for the Rotterdam Study is supported by the Netherlands Organization of Scientific Research NWO Investments (nr. 175.010.2005.011, 911-03-012). The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE and RIDE2), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), the Municipality of Rotterdam and the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) project nr. 050-060-810. We thank Pascal Arp, Mila Jhamai, Dr Michael Moorhouse, Marijn Verkerk and Sander Bervoets for their help in creating the GWAS database. We are grateful to the study participants, the staff from the Rotterdam Study and the participating general practioners and pharmacists. For the Australian cohort, we thank the Australian National Health and Medical Research Council (NHMRC; Grants 241944, 339462, 389927, 389875, 389891, 389892, 389938, 443036, 442915, 442981, 496739, 552485 and 552498) and the Australian Research Council (A7960034, A79906588, A79801419, DP0212016 and DP0343921) for funding. GWM and DRN are supported by the NHMRC Fellowship and the Australian Research Council Future Fellowship Schemes. We thank N Martin, P Visscher, D Duffy, A Henders, B Usher, E Souzeau, A Kuot, A McMellon, MJ Wright, MJ Campbell, A Caracella, L Bowdler, S Smith, S Gordon, B Haddon, D Smyth, H Beeby, O Zheng and B Chapman for their input into project management, databases, phenotype collection, and sample collection, processing and genotyping.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ligthart, L., de Vries, B., Smith, A. et al. Meta-analysis of genome-wide association for migraine in six population-based European cohorts. Eur J Hum Genet 19, 901–907 (2011). https://doi.org/10.1038/ejhg.2011.48
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.48
Keywords
This article is cited by
-
Genetics of migraine: where are we now?
The Journal of Headache and Pain (2023)
-
A Meta-Analysis of the Genome-Wide Association Studies on Two Genetically Correlated Phenotypes Suggests Four New Risk Loci for Headaches
Phenomics (2023)
-
Expression patterns of AEG-1 in the normal brain
Brain Structure and Function (2023)
-
Current Update on Categorization of Migraine Subtypes on the Basis of Genetic Variation: a Systematic Review
Molecular Neurobiology (2023)
-
Role of Omics in Migraine Research and Management: A Narrative Review
Molecular Neurobiology (2022)
