
Abstract
Migraine affects over a billion individuals worldwide but its genetic underpinning remains largely unknown. Here, we performed a genome-wide association study of 102,084 migraine cases and 771,257 controls and identified 123 loci, of which 86 are previously unknown. These loci provide an opportunity to evaluate shared and distinct genetic components in the two main migraine subtypes: migraine with aura and migraine without aura. Stratification of the risk loci using 29,679 cases with subtype information indicated three risk variants that seem specific for migraine with aura (in HMOX2, CACNA1A and MPPED2), two that seem specific for migraine without aura (near SPINK2 and near FECH) and nine that increase susceptibility for migraine regardless of subtype. The new risk loci include genes encoding recent migraine-specific drug targets, namely calcitonin gene-related peptide (CALCA/CALCB) and serotonin 1F receptor (HTR1F). Overall, genomic annotations among migraine-associated variants were enriched in both vascular and central nervous system tissue/cell types, supporting unequivocally that neurovascular mechanisms underlie migraine pathophysiology.
Similar content being viewed by others

Main
Migraine is a highly prevalent brain disorder characterized by disabling attacks of moderate-to-severe pulsating and usually one-sided headache that may be aggravated by physical activity, and can be associated with symptoms such as a hypersensitivity to light and sound, nausea and vomiting1. Migraine has a lifetime prevalence of 15–20% and is ranked as the second most disabling condition in terms of years lived with disability2,3. Migraine is three times more prevalent in females than in males. For about one-third of patients, migraine attacks often include an aura phase4 characterized by transient neurological symptoms such as scintillations. Hence, the two main migraine subtypes are defined as migraine with aura (MA) and migraine without aura (MO).
It has been debated for decades whether or not the migraine subtypes are in fact two separate disorders5,6,7, and, if so, what the underlying causes are. Prevailing theories about migraine pathophysiology emphasize neuronal and/or vascular dysfunction8,9. Current knowledge of disease mechanisms comes largely from studies of a rare monogenic subform of MA—familial hemiplegic migraine—for which three ion transporter genes (CACNA1A, ATP1A2 and SCN1A) have been identified10. The common forms of migraine (MA and MO) instead have a complex polygenic architecture with an increased familial relative risk5, increased concordance in monozygotic twins11 and a heritability of 40–60%12. The largest genome-wide association study (GWAS) thus far, with 59,674 cases and 316,078 controls, reported 38 genomic loci that confer migraine risk13. Subsequent analyses of these GWAS data showed enrichment of migraine signals near activating histone marks specific to cardiovascular and central nervous system (CNS) tissues14, as well as for genes expressed in vascular and smooth muscle tissues13. Other smaller GWAS15,16,17,18,19,20,21 have suggested ten additional loci. Of note, the previous datasets were too small to perform a meaningful comparison of the genetic background between migraine subtypes.
As migraine is globally the second largest contributor to years lived with disability2,3, there is clearly a large need for new treatments. Triptans, that is, serotonin 5-HT1B/1D receptor agonists, are migraine-specific acute treatments for the headache phase but are not effective in every patient, whereas preventive medication is far from satisfactory22. Recent promising alternatives for acute treatment are serotonin 5-HT1F receptor agonists (‘ditans’)23 and small-molecule calcitonin gene-related peptide (CGRP) receptor antagonists (‘gepants’)24,25. For preventive treatment, monoclonal antibodies (mAbs) targeting CGRP or its receptor have recently proven effective26, and new gepants for migraine prevention are under development27. Still, there remains an urgent need for treatment options for patients who do not respond to existing treatments. Genetics has proven promising in developing new therapeutic hypotheses in other prevalent complex diseases, such as cardiovascular disease28 and type 2 diabetes29, and we anticipate that large genetic studies of migraine could also yield similar insights.
We conducted a GWAS meta-analysis of migraine by adding to the previous meta-analysis13 42,410 new migraine cases from four study collections (Table 1). This increased the number of migraine cases by 71% for a total sample of 102,084 cases and 771,257 controls. Furthermore, we assessed the subtype specificity of the risk loci in 8,292 new MA and 6,707 new MO cases in addition to the 6,332 MA and 8,348 MO cases used previously13 (Table 2). Here, we report 123 genomic loci, of which 86 are previously unknown, and include the first four loci that reach genome-wide significance (P < 5 × 10−8) in MA. Our subtype data show compellingly that migraine risk is conferred both by risk loci that seem specific for only one subtype as well as by loci that are shared by both subtypes. Our findings also include new risk loci containing target genes of recent migraine drugs acting on the CGRP pathway and the serotonin 5-HT1F receptor. Finally, our data support the concept that migraine is brought about by both neuronal and vascular genetic factors, strengthening the view that migraine is truly a neurovascular disorder.
Results
Genome-wide meta-analysis
We combined data on 873,341 individuals of European ancestry (102,084 cases and 771,257 controls) from five study collections (Table 1 and Supplementary Table 1) and analyzed 10,843,197 common variants (Methods). Despite different approaches to the ascertainment of migraine cases across studies, pairwise genetic correlations were all near 1 (Supplementary Table 2), as determined by linkage disequilibrium (LD) score (LDSC) regression30, showing high genetic and phenotypic similarity across the studies, justifying their meta-analysis. Pairwise LDSC intercepts were all near 0, indicating little or no sample overlap (Supplementary Table 2).
The genomic inflation factor (λGC) of the fixed-effect meta-analysis results was 1.33 (Supplementary Fig. 1), which is in line with other large meta-analyses31,32,33 and is as expected for a polygenic trait34. The univariate LDSC35 intercept was 1.05 (s.e. 0.01), which, being close to 1.0, suggests that most of the genome-wide elevation of the association statistics comes from true additive polygenic effects rather than from a confounding bias such as population stratification. The LDSC analysis showed a linear trend between the variant’s LD score and its association with migraine, as expected from a highly polygenic phenotype such as migraine (Supplementary Fig. 2). The SNP heritability estimate from LDSC was 11.2% (95% confidence interval (CI) 10.8–11.6%) on a liability scale when assuming a population prevalence of 16%.
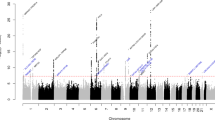
We identified 8,117 genome-wide significant (GWS; P < 5 × 10−8) variants represented by 170 LD-independent index variants (r2 < 0.1). We defined the risk loci by including all variants in high LD (r2 > 0.6) with the index variants and merged loci that were closer than 250 kb (Methods). This resulted in 123 independent risk loci (Fig. 1, Supplementary Table 3a and Supplementary Data 1 and 2). Of the 123 loci, 86 are previously unknown, whereas 36 overlap with the previously reported 47 autosomal risk loci (Supplementary Table 4) and one with the previously reported X chromosome risk locus. Of the 11 previously reported migraine risk loci that were not GWS in our study, 6 were GWS in Gormley et al.13 and had P < 3.50 × 10−5 in our data, 1 had P = 2.37 × 10−3, 3 had P > 0.14 and 1 was not available in our data (Supplementary Data 3). When we represented each risk locus by its lead variant, that is, the variant with the smallest P value, 47 GWS variants were LD-independent (r2 < 0.1) of the 123 lead variants, and, with a more stringent threshold (r2 < 0.01), 15 GWS variants remained LD independent of the 123 lead variants (Supplementary Table 5).

On the x axis, variants are plotted along the 22 autosomes and the X chromosome. The y axis shows the statistical strength of the association from the inverse–variance weighted fixed-effect meta-analysis as the negative log10 of the uncorrected two-sided P value (P). The horizontal line is the genome-wide significance threshold (P = 5 × 10−8). The 123 risk loci passing the threshold are divided into 86 new loci (purple) and 37 previously known loci (green). Adjacent chromosomes are colored in different shades of blue. Plotted are variants with P < 0.001.
In addition, we conducted an approximate stepwise conditional analysis for the 123 risk loci (Methods). Since sample sizes per variant varied considerably, we restricted the conditional analysis to variants with similar effective sample sizes to the lead variant. The conditional analysis returned six single nucleotide polymorphisms (SNPs) within the 123 risk loci that remained GWS after conditioning on the lead variants (Supplementary Table 6a,b).
Characterization of migraine risk loci
We mapped the 123 risk loci to genes by their physical location using the Ensembl Variant Effect Predictor (VEP)36. Of the lead variants, 59% (72/123) were within a transcript of a protein-coding gene, and 80% (99/123) of the loci contained at least one protein-coding gene within 20 kb, and 93% (114/123) within 250 kb (Supplementary Table 3). Of the 123 lead variants, 5 were missense variants (in genes PLCE1, MRGPRE, SERPINA1, ZBTB4 and ZNF462), and 40 more missense variants were in high LD (r2 > 0.6) with the lead variants (Supplementary Table 7a). Of note, three variants with a predicted high impact consequence on protein function were in high LD with the lead variants: (1) a stop gained variant (rs34358) with lead variant rs42854 (r2 = 0.85) in gene ANKDD1B, (2) a splice donor variant (rs66880209) with lead variant rs1472662 (r2 = 0.71) in RP11-420K8.1 and (3) a splice acceptor variant (rs11042902) with lead variant rs4910165 (r2 = 0.69) in MRVI1 (Supplementary Table 7b).
We used stratified LDSC (S-LDSC) to partition migraine heritability by 24 functional genomic annotations37,38. We observed enrichment for ten categories (Supplementary Fig. 3 and Supplementary Table 8), with conserved regions showing the highest enrichment (11.2-fold; P = 1.95 × 10−10), followed by coding regions (8.1-fold; P = 1.36 × 10−3) and enhancers (4.2-fold; P = 3.64 × 10−4).
Prioritization of candidate genes
We mapped the 123 lead variants to genes via expression quantitative trait locus (eQTL) association using GTEx v.8 (ref. 39) and data repositories included in FUMA40 at a false discovery rate (FDR) of 5% (Methods). The lead variants were cis-eQTLs for 589 genes (Supplementary Table 9), and variants in high LD with the lead variants were cis-eQTLs for an additional 624 genes (Supplementary Table 10). In total, 84% (103/123) of lead variants were cis-eQTLs for at least one gene. Tibial artery had the highest number (47/123) of lead variants as cis-eQTLs in GTEx v.8, and it was the only tissue type where the enrichment was statistically higher (P = 6.37 × 10−6) than expected based on the overall number of cis-eQTLs per tissue reported by GTEx (Supplementary Fig. 4 and Supplementary Note).
To prioritize candidate genes for the risk loci, we applied two approaches based on GTEx v.8 expression data: fine-mapping of causal gene-sets by FOCUS41 (Supplementary Table 11a) and a transcriptome-wide association study (TWAS) by S-PrediXcan42 combined with colocalization analysis using COLOC43 (Supplementary Table 11b).
With posterior probability (PP) > 0.5, FOCUS found candidate genes for 82 loci and S-PrediXcan + COLOC supported colocalization for 52 loci (Supplementary Table 11c). In total, 73 genes in 46 loci were prioritized by both methods. MRC2 and PHACTR1 were the only genes that both methods prioritized with strong evidence (PP > 0.99 for same tissue) and without any other gene prioritized within their loci.
Two of the new risk loci contain genes (CALCA/CALCB and HTR1F) whose protein products are closely related to targets of two migraine-specific drug therapies44. We observe a convincing association at the chromosome 11 locus that contains the CALCA and CALCB genes encoding CGRP itself (lead SNP rs1003194, P = 2.43 × 10−10; Fig. 2a), while none of the genes encoding CGRP receptor proteins (CALCRL, RAMP1 or RCP) show a statistically comparable association (all P > 10−4; Supplementary Fig. 5). Variant rs1003194 is a cis-eQTL for CALCB, but also for COPB1, PDE3B and INSC (Supplementary Table 9) and FOCUS prioritizes CALCA, CALCB and INSC (Supplementary Table 11c). In addition, a new locus on chromosome 3 contains HTR1F (lead SNP rs6795209, P = 1.23 × 10−8; Fig. 2b), which encodes the serotonin 5-HT1F receptor. Variant rs6795209 is a significant cis-eQTL for HTR1F, as well as for three other genes (CGGBP1, ZNF654, C3orf38) in the same locus (Supplementary Table 9). FOCUS or S-PrediXcan + COLOC did not prioritize HTR1F based on gene expression data (Supplementary Table 11c).

a, Locus containing CALCA and CALCB genes, encoding CGRP, which is the target of preventive and acute therapies via monoclonal antibodies and gepants. b, Locus containing the HTR1F gene, which encodes a serotonin 5-HT1F receptor that is the target of acute therapies via ditans. The x axis shows the chromosomal location, and the y axis shows the uncorrected two-sided negative log10 (P) from the inverse–variance weighted fixed-effects meta-analysis with 102,084 cases and 771,257 controls. The squared correlation to the lead variant is shown by colors based on the UK Biobank data for variants that have an effective sample size ±20% of the lead variant’s effective sample size. Horizontal line corresponds to P = 5 × 10−8. Blue graph shows the recombination rate.
Migraine subtypes with aura and without aura
Previously, Gormley et al.13 conducted subtype-specific GWAS with 6,332 MA cases against 144,883 controls and 8,348 MO cases against 139,622 controls, and reported that seven loci were GWS in MO but none were GWS in MA. Here, we added to the previous data 8,292 new MA and 6,707 new MO cases from headache specialist centers in Denmark and the Netherlands as well as from study collections in Iceland and UK Biobank (Table 2), for total sample sizes of 14,624 MA cases and 703,852 controls, and 15,055 MO cases and 682,301 controls. We estimated the effect size for each subtype at the 123 lead variants of the migraine GWAS (Supplementary Table 3b,c and Supplementary Data 4 and 5) and detected four GWS variants in the MA meta-analysis and 15 GWS variants in the MO meta-analysis. We also estimated a probability that the lead variant is either subtype-specific (that is, associated only with MO or with MA but not with both), shared by both subtypes, or not associated with either subtype (Methods; Supplementary Table 12a and Supplementary Data 6). With a probability above 95%, three lead variants (that is, rs12598836 in the HMOX2 locus, rs10405121 in the CACNA1A locus and rs11031122 in the MPPED2 locus) are MA-specific, while two lead variants (that is, rs7684253 in the locus near SPINK2 and rs8087942 in the locus near FECH) are MO-specific at a similar threshold. Nine lead variants were shared by MA and MO with >95% probability (Fig. 3a). In addition to the five subtype-specific lead variants, four other lead variants also showed differences in effect size between the subtypes (P < 0.05/123) (Fig. 3b).

a, Axes show the negative log10 (P) of MO (x axis; n = 697,356, 15,055 MO cases and 682,301 controls) and MA (y axis; n = 718,476, 14,624 MA cases and 703,852 controls) analyses. Two-sided P values are derived from inverse–variance weighted fixed-effect meta-analyses and are uncorrected. Symbols that are colored and annotated indicate >95% posterior probability that a nonzero effect is present in both MO and MA (model BOTH), or that the effect is present only in MO or only in MA but not both (models MO and MA, respectively). Variants with a probability <95% for each of the three models are shown as gray. Dashed lines show the genome-wide significance threshold (P = 5 × 10−8). b, Axes show logarithm of odds ratios (OR) for MO (x axis; n = 697,356, 15,055 MO cases and 682,301 controls) and MA (y axis; n = 718,476, 14,624 MA cases and 703,852 controls) calculated for the migraine risk allele. The effects at variants that have been colored and annotated differ between the subtypes at significance level of 0.0004 = 0.05/123. The 95% CIs for the logarithm of odds ratios are shown for the annotated variants. Dashed lines show the coordinate axes and the diagonal.
Phenome-wide association scans with National Human Genome Research Institute GWAS Catalog and FinnGen R4
Next, we conducted phenome-wide association scans (PheWAS) for the lead variants for 4,314 traits with reported associations in the National Human Genome Research Institute (NHGRI) GWAS catalog (https://www.ebi.ac.uk/gwas/) and for the GWAS summary statistics of 2,263 disease traits in the FinnGen release 4 data. We identified 25 lead variants that were reported to be associated with 23 different phenotype categories (Methods) in the GWAS Catalog, and 17 lead variants with 26 defined disease categories in FinnGen at P < 1 × 10−5. The categories with the highest number of reported associations were cardiovascular disease (7 lead variants) and blood pressure (6 lead variants) in the GWAS catalog, and diseases of the circulatory system (11 lead variants) in FinnGen. When we performed PheWAS for all variants in high LD (r2 > 0.6) with the lead variants, we observed associations for 79 loci with 54 different phenotype categories in the GWAS Catalog, and for 41 loci with 26 disease categories in FinnGen (Supplementary Table 13a and Supplementary Fig. 6).
These findings are consistent with previous results that migraine is a risk factor for several cardiovascular traits45,46,47, and genetically correlated with blood pressure48,49. However, we did not observe a trend in the direction of the allelic effects between migraine and coronary artery disease (CAD) or migraine and blood pressure traits (Supplementary Table 13d) using the latest meta-analysis of the CARDIoGRAMplusCD4 Consortium50 (n = 336,924) and blood pressure GWAS from UK Biobank51 (n = 422,771).
Enrichment in tissue or cell types and gene sets
We used LDSC applied to specifically expressed genes (LDSC-SEG)14 (Methods) to evaluate whether the polygenic migraine signal was enriched near genes that were particularly active in certain tissue or cell types as determined by gene expression or activating histone marks. Using multitissue gene expression data, we found enrichment at FDR 5% in three cardiovascular tissue/cell types, that is, aorta artery (P = 1.78 × 10−4), tibial artery (P = 3.60 × 10−4) and coronary artery (P = 4.29 × 10−4) (Table 3 and Supplementary Table 14a), all of which have previously been reported enriched in migraine without aura14. The fine-scale brain expression data from GTEx, since recently including 13 brain regions, showed enrichment in the caudate nucleus of striatum—a component of basal ganglia (P = 6.02 × 10−4; Table 3 and Supplementary Table 14b). With chromatin-based annotations, we found enrichment in five CNS cell types, three cardiovascular cell types, one cell type of the digestive system, one musculoskeletal/connective cell type and ovary tissue (Table 3 and Supplementary Table 14c). In addition to replicating previous findings13,14, the signal linking to ovary tissue has not been reported before.
Finally, we used DEPICT52 to identify tissues whose eQTLs were enriched for migraine-associated variants. The tissue enrichment analysis replicated three previously reported tissues13: arteries (nominal P = 1.03 × 10−3), stomach (nominal P = 1.04 × 10−3) and upper gastrointestinal tract (nominal P = 1.29 × 10−3) (Supplementary Table 14a). Results of gene-set analyses using DEPICT52 and MAGMA53 are presented in Supplementary Tables 15 and 16.
Discussion
We conducted the largest GWAS meta-analysis on migraine thus far by combining genetic data on 102,084 cases and 771,257 controls. We identified 123 migraine risk loci, of which 86 are previously unknown since the previous migraine meta-analysis, which yielded 38 loci13. This shows that we have now reached the statistical power for rapid accumulation of new risk loci for migraine, in line with the progress of GWAS seen with other common diseases54, and as expected for a highly polygenic disorder like migraine55.
Migraine subtypes MO and MA were defined as separate disease entities some 30 years ago, and, since then, the debate has continued as to what extent they are biologically similar. Over the years, arguments in favor6 and against5 have been presented, but convincing genetic evidence to support subtype-specific risk alleles has been lacking in genetic studies with smaller sample sizes18,56,57. Here, we increased considerably the evidence for subtype specificity of some risk alleles by including new migraine subtype data at the 123 migraine risk variants. We observed that, with a probability of >95%, three lead variants (in HMOX2, in CACNA1A and in MPPED2) are associated with MA but not MO. Of these variants, CACNA1A is a well-known gene linked to familial hemiplegic migraine, a rare subform of MA58,59. The observation that CACNA1A seems involved in both monogenic and polygenic forms of migraine provides a gene-based support for the increased sharing of common variants between the two disorders55. We find no evidence that any of the seven loci, previously reported as GWS in MO but not in MA13, would be specific for MO, while four of them (LRP1, FHL5, near FGF6 and near TRPM8) are among the nine loci shared by both subtypes with a probability over 95%. Loci (for example, LRP1 and FHL5) that are strongly associated with both subtypes provide convincing evidence for a previous hypothesis that the subtypes partly share a genetic background13,60. In accordance with our analysis, effects in both subtypes were suggested before at the TRPM8 and TSPAN2 loci, whereas, in contrast to our results, the LRP1 locus was previously reported to be specific for MO56. Finally, we also detected four lead variants (including LRP1) that do not seem specific for MO but do confer a higher risk for MO than for MA.
It has been long debated whether migraine has a vascular or a neuronal origin, or whether it is a combination of both8,9,61,62. Here, we found genetic evidence for the role of both vascular and central nervous tissue types in migraine from several tissue enrichment analyses, which refined earlier analyses based on smaller sample sizes13,14.
With respect to a vascular involvement in the pathophysiology of migraine, both gene expression and chromatin annotation data from LDSC-SEG showed that migraine signals are enriched for genes and cell-type-specific annotations that are highly expressed in aorta and tibial and coronary arteries. The involvement of arteries was also proposed by our DEPICT tissue enrichment analysis. In addition, cardiovascular disease and blood pressure phenotypes were among the top categories in the PheWAS analyses. These results are consistent with previous reports of a shared etiology and some genetic correlation between migraine and cardiovascular and cerebrovascular endpoints47,48,49,63,64,65,66,67. However, in our analysis, the migraine risk alleles neither consistently increased nor consistently decreased the risk of CAD or the risk of hypertension.
A key role of the CNS in migraine pathophysiology has emerged from animal models, human imaging and neurophysiological studies10,68, while support for CNS involvement from genetic studies has been more difficult to obtain. A likely reason is the paucity of gene expression data from CNS tissue types, but recently more data have become available, making such studies feasible. Our LDSC-SEG analysis using gene expression data from 13 brain regions showed an enrichment for caudate nucleus in the basal ganglia, and with chromatin-based annotations for five CNS tissue types: dorsolateral prefrontal cortex, neurospheres derived from cortex, fetal brain, germinal matrix and neurospheres derived from ganglion eminence. Alterations in the structure and/or function of several brain regions68,69,70, including basal ganglia, cortex, hypothalamus, thalamus, brainstem, amygdala and cerebellum, have been reported for individuals who suffer from migraine, but the cause of these changes is not known.
In addition to the support for the hypothesis that both vascular system and CNS are important in migraine pathogenesis8,68,71, the tissue enrichment analyses also reported some tissue types of the digestive system as well as ovary at FDR 5%. Given the female preponderance and suggested influence of sex hormones (for example, menstrual-related migraine) in migraine72,73,74, the involvement of the ovary is an interesting finding, although the statistical evidence for it remains weaker at present compared with that for the vascular system and CNS.
A particularly interesting finding in our GWAS was the identification of risk loci containing genes that encode targets for migraine-specific therapeutics. One new locus contains the CALCA and CALCB genes on chromosome 11 that encode calcitonin gene-related peptide (CGRP). CGRP-related monoclonal antibodies have been successful for the preventive treatment of migraine75, and they are considered as a major breakthrough in migraine-specific treatments since the development of the triptans for acute migraine over two decades ago. Another new locus contains the HTR1F gene that encodes serotonin 5-HT1F receptor, which is the target of another recent migraine drug class called ditans76. Ditans provide a promising acute treatment, especially for those migraine patients that cannot use triptans because of cardiovascular risk factors23. These two new GWAS associations near genes that are already targeted by effective migraine drugs suggest that there could be other potential drug targets among the new loci, and provide a clear rationale for future GWAS efforts to increase the number of loci by increasing sample sizes further. In addition, GWAS data with migraine subtype information can help prioritize treatment targets for particular migraine symptomatology, such as aura symptoms, that lack treatment options at present. More generally, using genetic evidence when selecting new drug targets is estimated to double the success rate in clinical development77,78.
Even though we observed links between our new risk loci and known target genes of effective migraine drugs, the accurate gene prioritization at risk loci remains challenging. First, robust fine-mapping would require accurate LD information79, which is typically lacking in meta-analyses and further distorted from reference panels by variation in effective sample size across variants. Second, computational approaches to gene prioritization require further methodological work80 and extension to additional sources of functional data to provide more robust and comprehensive gene prioritization results. Another limitation of our study is that a large proportion of migraine diagnoses are self-reported. Therefore, we cannot rule out misdiagnosis, such as, for example, tension headache being reported as migraine, which could overemphasize genetic factors related to general pain mechanisms and not migraine per se. Regardless, the high genetic correlation that we observed supports a strong phenotypic concordance between the study collections that also included deeply phenotyped clinical cohorts from headache specialist centers, which were instrumental for the migraine subtype analyses. While the subtype data provided convincing evidence of both loci with genetic differences and other loci with genetic overlap between subtypes, larger samples are still needed to achieve a more accurate picture of the similarities and differences in genetic architecture behind the subtypes.
To conclude, we report the largest GWAS meta-analysis of migraine so far, detecting 123 risk loci. We demonstrated that both the vascular system and CNS are involved in migraine pathophysiology, supporting the notion that migraine is a neurovascular disease. Our subtype analysis of migraine with aura and migraine without aura shows that these migraine subtypes have both shared risk alleles and risk alleles that seem specific to one subtype. In addition, new loci include two targets of recently developed and effective migraine treatments. Therefore, we expect that these and future GWAS data will reveal more of the heterogeneous biology of migraine and potentially point to new therapies against migraine—a leading burden for population health throughout the world.
Methods
Cohorts and phenotyping
All participating studies were approved by local research ethics committees, and written informed consent was obtained from all study participants. For all the participating studies, an approval was received to use the data in the present work. Study-specific ethics statements are provided in the Supplementary Note.
First, we performed a genome-wide meta-analysis on migraine including five study collections, as listed in Table 1 and Supplementary Table 1. Second, we performed subtype-specific meta-analyses on MA and on MO, both including five study collections listed in Table 2, for the 123 independent risk variants identified in the migraine analysis. A description of the study collections is given in the Supplementary Note. In particular, the migraine phenotype has been self-reported in other cohorts except in IHGC2016, where a subset of patients were phenotyped in specialized headache centers, as previously explained13.
Quality control
Before the meta-analysis, a standard quality control (QC) protocol was applied to each individual GWAS. Related individuals were removed from all other cohorts except HUNT (which modeled relatedness via a logistic mixed model) by using an identity by descent cut-off of 0.185 or smaller. Multiallelic variants were excluded from all studies, and only variants that satisfied the following thresholds were kept for further analysis: minor allele frequency (MAF) > 0.01, IMPUTE2 info or MACH r2 > 0.6 and, when available, Hardy-Weinberg equilibrium (HWE) P value >1 × 10−6 and missingness <0.05. Variants were matched by chromosome, position and alleles to the UK Biobank data. Indels were recoded as insertions (I) and deletions (D). For each study, SNPs with an effect allele frequency (EAF) discrepancy of >0.30 and indels with EAF discrepancy of >0.20 to UK Biobank were excluded. MAF and EAF plots of cohorts against the reference cohort are shown in Supplementary Data 7. We conducted a sensitivity analysis on strand-ambiguous SNPs (with alleles A/T or G/C), by counting, for each pair of studies, how often the same allele of A/T or G/C SNP was coded as the minor allele in both cohorts, as a function of MAF threshold (Supplementary Table 17). Minor alleles were same at least in 97.39% of the SNPs without MAF threshold and the corresponding proportions were 99.96% and 79.58% when MAF < 0.25 and when MAF > 0.4, respectively. The very high concordance for SNPs with MAF < 0.25 suggests that the strand-ambiguous SNPs were labeled consistently for almost every SNP. Therefore, we did not exclude any SNPs based on possible labeling mismatches due to strand ambiguity.
Statistical analysis
All statistical tests conducted were two-sided unless otherwise indicated. The GWAS for the individual study cohorts were performed by logistic regression with an additive model of imputed dosage of the effect allele on the log-odds of migraine. The analyses for IHGC2016 (ref. 13) and 23andMe19 have been described before. For UKBB data and GeneRISK data, we used PLINK v.2.0 (ref. 81). For HUNT data, we used a logistic mixed model with the saddlepoint approximation as implemented in SAIGE v.0.20 (ref. 82) that accounts for the genetic relatedness. All models were adjusted for sex and at least for the four leading principal components of the genetic population structure (Supplementary Table 18). Age was used as a covariate when available. A detailed description is provided in Supplementary Note. For the chromosome X meta-analysis, male genotypes were coded as {0,2} in all cohorts, and the GWAS were conducted with an X chromosome inactivation model that treats hemizygous males as equivalent to homozygous females83.
We performed an inverse–variance weighted fixed-effect meta-analysis on the five study collections by using GWAMA84. After the meta-analysis, we excluded the variants with effective sample size Neff < 5,000 to remove results with very low precision compared with most variants and were left with 10,843,197 variants surpassing the QC thresholds. We estimated the effective sample size for variant i as
where fi is the effect allele frequency for variant i and s.e.i is the s.e. for variant i estimated by the GWAS software. This quantity approximates the value 2 N t(1 – t)I, where N is the total sample size (cases + controls), t is the proportion of cases and I is the imputation info (derivation in Supplementary Note).
Risk loci
There were 8,117 GWS variants with the meta-analysis P value <5 × 10−8. For 8,067 of them that were available in UK Biobank, an LD matrix was obtained from UK Biobank using a random sample of 10,000 individuals included in the UKBB GWAS. We defined the index variants as the LD-independent GWS variants at LD threshold of r2 < 0.1 in the following way. First, the GWS variant with the lowest P value was chosen and, subsequently, all GWS variants that were in LD with the chosen variant (r2 > 0.1) were excluded. Next, out of the remaining GWS variants, the variant with the lowest P value was chosen and the GWS variants in LD with that variant were excluded. This procedure was repeated until there were no GWS variants left. Out of the 8,067 variants with LD information, 170 were LD-independent (at r2 < 0.1). For 18/50 variants that were not found in UK Biobank, LD information was available from the 23andMe data, and all 18 variants were in LD (r2 > 0.1) with some index variant. Of the 18 variants, 2 (rs111404218 and rs12149936) had lower P value than the original index variant they were in LD with and, hence, they replaced the original index variants. For 32 GWS variants, LD remained unknown. Thus, at this stage, the GWS associations were represented by 202 = 168 + 2 + 32 index variants.
Next, to define the risk loci and their lead variants, an LD block around each index variant was formed by the interval spanning all GWS variants that were in high LD (r2 > 0.6) with the index variant. Sizes of these regions ranged from 1 bp (only the variant itself, for example, the variants with unknown LD) to 1,089 kb. Sets of regions that were less than 250 kb away from each other were merged (distance from the end of the first region to the beginning of the second region). This definition resulted in 126 loci. All other GWS variants were included in their nearest locus based on their position and the locus boundaries were updated and, finally, loci within 250 kb from each other were merged. This resulted in our final list of 123 risk loci. Each risk locus was represented by its lead variant defined as the variant with the lowest P value and named by the nearest protein-coding gene to the lead variant or by the nearest noncoding gene if there was no protein-coding gene within 250 kb. The term ‘Near’ was added to the locus name if the lead variant did not overlap with a gene transcript. We note that the nearest gene to the lead variant need not be a causal gene. None of the 32 variants without LD information became a lead variant of a risk locus because all had a variant in the vicinity with a smaller P value.
We annotated and mapped these loci by their physical position to genes by using the Ensembl Variant Effect Predictor (VEP, GRCh37)36. We used two different thresholds for annotating the nearest genes: a distance of 20 kb and 250 kb to the nearest transcript of a gene. The filtered results including all variants within a gene or a regulatory element are presented in Supplementary Table 7b.
Stepwise conditional analysis
We performed a stepwise conditional analysis (CA) on each risk locus by using FINEMAP v.1.4 (ref. 85). FINEMAP uses GWAS summary statistics together with an LD reference panel and does not require individual-level data. When the reference LD does not accurately match the GWAS data, full fine-mapping is prone to false positives79. A simpler stepwise CA is more robust to inaccuracy in reference LD because CA has a much smaller search space than full fine-mapping, and therefore CA is less likely to run into most problematic variant combinations where LD is very inaccurate. Since we did not have the full in-sample LD from our GWAS data, we carried out only the CA and not the full fine-mapping. For the CA, we included only the SNPs, but no indels, and we used the same reference LD from the UK Biobank data as we used to define the risk loci. We restricted the CA only to the variants with a similar effective sample size (Neff) by using a threshold of ±10% of the Neff of the lead SNP of the risk locus, because our summary statistics came from the meta-analysis where sample sizes per variant vary greatly. This filter excluded approximately 17% of all GWS variants and was necessary since otherwise CA led to spurious conditional P values, such as P < 10−250, for some loci. Consequently, for two of the loci where the lead variant was an indel, the lead variant was not included in the CA. For such regions, we checked that the new lead variant from the CA output was in LD (r2 > 0.3) with the original lead variant. For one locus (rs111404218) where the lead variant does not have LD information in the UK Biobank data, there were no GWS variants left in the CA after filtering by Neff. We used the standard GWS (P < 5 × 10−8) threshold to define the secondary variants that were conditionally independent from the lead variant. The CA results are in Supplementary Tables 6a,b.
eQTL mapping to genes and tissues
We used two data sources to map the risk variants to genes via eQTL associations. From the GTEx v.8 database (https://gtexportal.org), we downloaded the data of 49 tissues. We first mapped all 123 lead variants to all significant cis-eQTLs across tissues using the FDR cut-off of 5% as provided by the GTEx project39. Next, we also mapped the variants in high LD (r2 > 0.6) with the lead variants to all significant cis-eQTLs. Finally, we filtered the results to include only the new significant gene-tissue pairs that were not implicated by the lead variants. Results are shown in Supplementary Tables 9 and 10.
With FUMA v.1.3.6 (ref. 40), we mapped the 123 lead variants, and the variants in high LD (r2 > 0.6) with the lead variants, to the other eQTL data repositories provided by FUMA except GTEx, that is, Blood eQTL Browser86, BIOS QTL browser87, BRAINEAC88, MuTHER89, xQTLServer90, CommonMind Consortium91, eQTLGen92, eQTL Cataloque93, DICE94, scRNA eQTLs95 and PsychENCODE96. Results are shown in Supplementary Tables 9 and 10.
To study whether the lead variants were enriched in any of the 49 tissues from GTEx v.8, we fitted a linear regression model where the number of lead variants that are significant cis-eQTLs for a specific tissue was used as the outcome, and the overall number of genes with at least one significant cis-eQTL reported by GTEx for the tissue was the predictor39. We did a separate regression model for each tissue type by leaving the tissue of interest out from the model, and we used the model fitted on the other tissues for predicting the outcome variable for the tissue type of interest. Finally, we checked in which tissues the true observed number of migraine lead variants was outside of the 95% prediction intervals as given by the function ‘predict.lm(‘interval=’prediction’)’ in R software. Details of the procedure are in the Supplementary Note.
LD-score regression
We estimated both the SNP heritability (\(h_{\mathrm{SNP}}^2\)) of migraine and pairwise genetic correlations (rG) between each pair of study collections using LDSC v.1.0.0 (refs. 30,35). SNP heritability and genetic correlations were estimated using European LD scores from the 1000 Genomes Project Phase 3 data for the HapMap3 SNPs, downloaded from https://data.broadinstitute.org/alkesgroup/LDSCORE/. We reformatted the meta-analysis association statistics to LDSC format with munge-tool, which excluded variants that did not match with the HapMap3 SNPs, had strand ambiguity (that is, A/T or G/C SNPs), MAF <0.01 or missingness more than two-thirds of the 90th percentile of the total sample size, or resided in long-range LD regions97, in centromere regions or in the major histocompatibility locus (MHC) of chromosome 6, leaving 1,165,201 SNPs for the LDSC analyses. We used a migraine population prevalence of 16% and a sample proportion of cases of 11.7% = 102,084/(102,084 + 771,257) to turn the LDSC slope into the estimate of \(h_{\mathrm{SNP}}^2\) on the liability scale98. Pairwise genetic correlation results are listed in Supplementary Table 2. We note that in the previous migraine meta-analysis13, LDSC reported \(h_{\mathrm{SNP}}^2\) value of 14.6% (13.8–15.5%), which was considerably larger than the value 11.2% (10.8–11.6%) that we report in our analysis. When we ran our LDSC pipeline on the data of Gormley et al.13, we estimated \(h_{\mathrm{SNP}}^2\) value of 10.6% (10.1–11.1%). Thus, it seems that our liability transformation estimates lower values of heritability than the transformation used by Gormley et al.13.
Stratified LD-score regression
We used S-LDSC to partition the SNP heritability by functional genomic annotations37. We used the baseline-LD model38 that contains 75 annotations, including conserved, coding and regulatory regions of the genome and different histone modifications. The baseline-LD model adjusts for MAF- and LD-related annotations, such as recombination rate and predicted allele age, which decreases the risk of model misspecification37,38,99. We used the same QC as with the univariate LDSC, and the baseline LDv.1.1 European LD scores estimated from the 1000 Genomes Project Phase 3, downloaded from https://data.broadinstitute.org/alkesgroup/LDSCORE/. We set the significance threshold for enrichment of individual binary functional annotations to α = 0.05/24, as we considered only 24 unique functional annotations without the flanking regions. Results are listed in Supplementary Table 8.
Subtype analyses of MA and MO
First, we combined new MA and MO data (Table 2) with the previously used migraine subtype-specific meta-analysis data13, and estimated migraine subtype-specific effect sizes for the 123 lead variants from the migraine meta-analysis. We tested how often the direction of allelic effects was similar between the IHGC MA/MO and the new cohorts using a binomial test (Supplementary Table 12b). Next, we stratified the lead variants by using the information from the migraine subtype-specific analyses. For each of the variants, we estimated probabilities between four possible explanations of the observed data that we call ‘NULL’, ‘MO’, ‘MA’ and ‘BOTH’. Under model NULL, the effect is not present in either of the migraine subtypes (that is, the effect is zero); under model MO or MA, the effect is present only in MO or only in MA but not in both; and under model BOTH, a nonzero effect is shared by both MO and MA. We used a Bayesian approach for model comparison that combines a bivariate Gaussian prior distribution on the two effect sizes with a bivariate Gaussian approximation to the likelihood using GWAS summary statistics100. Across all models, the prior s.d. for the effect is 0.2 on the log-odds scale for nonzero effects and 0 for a zero effect. The bivariate priors for the four models are as follows: NULL assumes a zero effect in both migraine subtypes, MO and MA assume a nonzero effect for one subtype and a zero effect for the other subtype, and BOTH combines the fixed-effect model (exactly the same effect in both subtypes) with the independent-effects model (the two effect sizes are nonzero but uncorrelated with each other) with equal weights. Finally, we assumed that each of the four models (NULL, MO, MA and BOTH) is equally probable a priori, which we considered an appropriate assumption since all these variants show a convincing association to overall migraine (P < 5 × 10−8). Then we used the Bayes formula to work out the posterior probability on each model. The results are shown in Fig. 3a, thresholded by a probability cut-off of 95%, and in Supplementary Table 12a. The correlation parameter between MO and MA GWAS statistics needed in the bivariate likelihood approximation was estimated to be 0.148 using the empirical Pearson correlation of the effect size estimates of the common variants (MAF > 0.05), which did not show a strong association to either of the migraine subtypes (P > 1 × 10−4)101. We tested whether the effect sizes between MA and MO were equal at a Bonferroni corrected significance threshold of α = 0.05/123 by using a normal approximation and accounting for the correlation in effect size estimators.
We note that the amount of information in the data (‘statistical power’) is taken into account automatically in this model comparison, which we consider an advantage compared with a comparison of the raw P values between the subtype analyses that does not automatically account for statistical power. In particular, observing a GWS P value (P < 5 × 10−8) in one subtype but not in the other subtype is not yet evidence for a subtype-specific locus, because the effect could still be nonzero also for the other subtype but simply lack power to reach the stringent GWS threshold. Finally, we point out that the inference in the model comparison approach is conditional on the particular set of models being included in the comparison, as well as on the particular choice of the prior distributions.
PheWAS with NHGRI GWAS catalog and FinnGen R4
We performed PheWAS for the 123 lead variants using the NHGRI GWAS catalog and the FinnGen R4 GWAS summary statistics. In addition, we performed the same lookups for the 123 risk loci including all variants in high LD (r2 > 0.6) with the lead variants. With the GWAS catalog, we first downloaded all the available results (4,314 traits) from the GWAS catalog webpage (accessed April 6, 2020). Next, we obtained all the associations for the 123 risk loci with all the high LD variants included using P value thresholds of P < 1 × 10−5, P < 1 × 10−6 and P < 1 × 10−4 (Supplementary Table 13a–c). Because the GWAS catalog includes results from several different GWAS for the same phenotype or for a very similar phenotype with a different name, we divided the phenotype associations into broader categories. The new categories are listed in Supplementary Table 19. The same approach was used for the PheWAS of FinnGen R4. We first downloaded all the available summary statistics (2,263 endpoints) and, next, obtained all the associations for the 123 risk loci using the same three P value thresholds as with the GWAS catalog (Supplementary Table 13a–c). We also divided similar endpoints into broader categories, which are listed in Supplementary Table 20.
We tested the direction of allelic effects between migraine and the following three traits that shared multiple associated variants with migraine: CAD50, diastolic blood pressure51 and systolic blood pressure51. We first took all migraine lead variants that were available also in the summary statistics of the other trait without any P value threshold and used a binomial test to test whether the proportion of variants with same direction of effects was 0.5. Next, we used a P value threshold of 1 × 10−5 for the association with the other trait. Results are in Supplementary Table 13d.
LD-score regression applied to specifically expressed genes
We used LDSC-SEG14 to identify tissues and cell types implicated by the migraine GWAS results. LDSC-SEG uses gene expression data and GWAS results from all variants together with an LD reference panel. For our analyses, we used the same QC as for the other LDSC analyses and six different sets of readily constructed annotation-specific LD scores downloaded from https://data.broadinstitute.org/alkesgroup/LDSCORE/LDSC_SEG_ldscores/: multitissue gene expression, multitissue chromatin, GTEx brain, Cahoy, Corces ATAC and ImmGen LD Scores. FDR was controlled by the Benjamini–Hochberg method. The results are in Supplementary Table 14a–f. There were no significant results with the Cahoy, Corces ATAC and ImmGen data at FDR 5%.
Multimarker analysis of genomic annotation
We applied multimarker analysis of genomic annotation (MAGMA) v.1.09 (ref. 53) to identify genes and gene sets associated with the migraine meta-analysis results. First, we mapped the meta-analysis SNPs to 18,985 protein-coding genes based on their physical position in the National Center for Biotechnology Information 37 build by using default settings of MAGMA. Next, we performed a gene-based analysis using the default SNPwise-mean model and the same UK Biobank LD reference as for the other analyses. We applied a Bonferroni correction (α = 0.05/18,985) to identify significantly associated genes for migraine with the results listed in Supplementary Table 16a. Finally, we used the results from the gene-based analysis to perform a gene-set analysis by using two different gene-set collections from the Molecular Signature Database v.7.0 (refs. 102,103): the curated gene sets containing 5,500 gene sets and the GO gene sets containing 9,988 gene sets. We performed the gene-set analysis using the competitive gene-set model and one-sided test that tests whether the genes in the gene-set are associated more strongly with the phenotype compared to the other genes. To correct for multiple testing, we used a Bonferroni correction (α = 0.05/(5,500 + 9,988)). Results are in Supplementary Table 16b,c and in Supplementary Fig. 7.
DEPICT
DEPICT52 is an integrative tool to identify the most likely causal genes at associated loci, and enriched pathways and tissues or cell types in which the genes from the associated loci are highly expressed. As an input, DEPICT takes a set of trait-associated SNPs. First, DEPICT uses coregulation data from 77,840 microarrays to predict biological functions of genes and to construct 14,461 reconstituted gene sets. Next, information of similar predicted gene functions is used to identify and prioritize gene sets that are enriched for genes in the associated loci. For the tissue- and cell-type-enrichment analysis, DEPICT uses a set of 37,427 human gene expression microarrays. We used DEPICT v.1.194 and ran the analyses twice for each of the P value thresholds for clumping, as recommended52, and using the default settings of 500 permutations for bias adjustment and 50 replications for the FDR estimation and for the P value calculation. As an input, we used only the autosomal SNPs and the same UK Biobank LD reference data as for the other analyses. First, we ran the analysis using a clumping P value threshold of 5 × 10−8 that resulted in 165 clumps formed from 7,672 variants (Supplementary Table 15d–f). Second, we used a P value threshold of 1 × 10−5 leading to 612 clumps formed from 22,480 variants (Supplementary Table 15a–c).
Transcriptome-wide association study and colocalization
We performed a transcriptome-wide association study (TWAS) by S-PrediXcan42 v.0.7.5 using GTEx v.8 multivariate adaptive shrinkage models (MASHR-M) for 49 tissues downloaded from predictdb.org and the European 1000 Genomes v.3 LD reference panel (hg38; https://zenodo.org/record/3657902/). We followed the recommended QC protocol, and first harmonized and imputed the migraine summary statistics to ensure an optimal overlap with the GTEx v.8 expression weights. After harmonization and summary statistic imputation, 8,909,736 variants were available for the TWAS. We performed the analysis with default settings to identify significant gene-tissue pairs. We applied a Bonferroni corrected significance level of α = 0.05/662,726, corresponding to the number of unique gene-tissue pairs tested.
Next, we performed colocalization analysis with COLOCv.4.0.4(ref. 43) R package for the 1,844 significant gene-tissue pairs to indicate pairs that could be due to LD contamination. COLOC compares five hypotheses where the null hypothesis (H0) corresponds to no association to either eQTL or GWAS, H1 and H2 correspond to associations with only one of the traits, H3 corresponds to association with both eQTL and GWAS but at distinct causal variants, and H4 corresponds to association with both eQTL and GWAS at a shared causal variant. We set a prior probability for colocalization as p12 = 5 × 10−6 for all tested regions and restricted the analysis to variants that had Neff ± 10% of the Neff of the lead variant of the region. Results are presented in Supplementary Table 11b.
Fine-mapping of causal gene sets
To prioritize genes for the migraine loci, we applied a gene-based fine-mapping approach using fine-mapping of causal gene sets (FOCUS) v.0.7 (ref. 41). FOCUS is a Bayesian approach that models predicted expression correlations among TWAS signals to estimate posterior probabilities for all genes within a tested region.
We used the European 1000 Genomes v.3 LD reference panel and same GTEx v.8 predicted expression weights for the 49 tissues as with S-PrediXcan. First, we mapped the migraine summary statistics from hg37 to hg38 with UCSC liftOver104. Next, we followed the suggested QC protocol and applied the modified munge-tool to obtain cleaned summary statistics. After the QC steps, we had 6,237,177 variants left for the analysis. We performed tissue-prioritized fine-mapping of gene-sets for the 49 tissues with otherwise default settings except that we increased the P value threshold to 1 × 10−4 so that the fine-mapping would cover most of the same regions that contained at least one significant gene-tissue pair by S-PrediXcan. Posterior inclusion probability (PIP) from FOCUS is reported for all available significant S-PrediXcan gene-tissue pairs in Supplementary Table 11b, and all prioritized genes by FOCUS with PIP >0.9 are reported in Supplementary Table 11a.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Results for 8,117 genome-wide significant SNP associations (P < 5 × 10−8) from the meta-analysis including 23andMe data are available on the International Headache Genetics Consortium website (http://www.headachegenetics.org/content/datasets-and-cohorts). Genome-wide summary statistics for the other study collections except 23andMe are available for bona fide researchers (contact Dale Nyholt, d.nyholt@qut.edu.au) within 2 weeks from the request. The full GWAS summary statistics for the 23andMe discovery data set will be made available through 23andMe to qualified researchers under an agreement with 23andMe that protects the privacy of the 23andMe participants. Please visit https://research.23andme.com/collaborate/#publication for more information and to apply to access the data.
Code availability
R code for the subtype specificity analysis: https://github.com/mjpirinen/migraine-meta.
References
Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition. Cephalalgia 38, 1–211 (2018).
Vos, T. et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396, 1204–1222 (2020).
Steiner, T. J. et al. Migraine remains second among the world’s causes of disability, and first among young women: findings from GBD2019. J. Headache Pain 21, 137 (2020).
Russell, M. B., Rasmussen, B. K., Thorvaldsen, P. E. R. & Olesen, J. E. S. Prevalence and sex-ratio of the subtypes of migraine. Int. J. Epidemiol. 24, 612–618 (1995).
Russell, M. B. & Olesen, J. Increased familial risk and evidence of genetic factor in migraine. BMJ 311, 541–544 (1995).
Kallela, M., Wessman, M., Havanka, H., Palotie, A. & Farkkila, M. Familial migraine with and without aura: clinical characteristics and co-occurrence. Eur. J. Neurol. 8, 441–449 (2001).
de Boer, I., van den Maagdenberg, A. M. J. M. & Terwindt, G. M. Advance in genetics of migraine. Curr. Opin. Neurol. 32, 413–421 (2019).
Tfelt‐Hansen, P. C. & Koehler, P. J. One hundred years of migraine research: major clinical and scientific observations from 1910 to 2010. Headache 51, 752–778 (2011).
Anttila, V., Wessman, M., Kallela, M. & Palotie, A. in Handbook of Clinical Neurology, Vol. 148 (eds Geschwind, D.H., Paulson, H.L. & Klein, C.) Ch. 31 (Elsevier, 2018).
Ferrari, M. D., Klever, R. R., Terwindt, G. M., Ayata, C. & van den Maagdenberg, A. M. J. M. Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol. 14, 65–80 (2015).
Ulrich, V., Gervil, M., Kyvik, K. O., Olesen, J. & Russell, M. B. Evidence of a genetic factor in migraine with aura: a population-based Danish twin study. Ann. Neurol. 45, 242–246 (1999).
Gervil, M., Ulrich, V., Kaprio, J., Olesen, J. & Russell, M. B. The relative role of genetic and environmental factors in migraine without aura. Neurology 53, 995–999 (1999).
Gormley, P. et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet. 48, 856–866 (2016).
Finucane, H. K. et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018).
Anttila, V. et al. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat. Genet. 42, 869–873 (2010).
Chasman, D. I. et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat. Genet. 43, 695–698 (2011).
Freilinger, T. et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat. Genet. 44, 777–782 (2012).
Anttila, V. et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat. Genet. 45, 912–917 (2013).
Pickrell, J. K. et al. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 48, 709–717 (2016).
Chen, S.-P. et al. Genome-wide association study identifies novel susceptibility loci for migraine in Han Chinese resided in Taiwan. Cephalalgia 38, 466–475 (2018).
Chang, X. et al. Common variants at 5q33.1 predispose to migraine in African-American children. J. Med. Genet. 55, 831–836 (2018).
Tfelt-Hansen, P. & Olesen, J. Taking the negative view of current migraine treatments. CNS Drugs 26, 375–382 (2012).
Kuca, B. et al. Lasmiditan is an effective acute treatment for migraine. Neurology 91, e2222–e2232 (2018).
Dodick, D. W. Migraine. Lancet 391, 1315–1330 (2018).
Lipton, R. B. et al. Effect of ubrogepant vs placebo on pain and the most bothersome associated symptom in the acute treatment of migraine: the ACHIEVE II randomized clinical trial. JAMA 322, 1887–1898 (2019).
Charles, A. & Pozo-Rosich, P. Targeting calcitonin gene-related peptide: a new era in migraine therapy. Lancet 394, 1765–1774 (2019).
Goadsby, P. J. et al. Safety, tolerability, and efficacy of orally administered atogepant for the prevention of episodic migraine in adults: a double-blind, randomised phase 2b/3 trial. Lancet Neurol. 19, 727–737 (2020).
Cohen, J. et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 37, 161–165 (2005).
Flannick, J. et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat. Genet. 46, 357–363 (2014).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Nagel, M. et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat. Genet. 50, 920–927 (2018).
Pardiñas, A. F. et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 50, 381–389 (2018).
Howard, D. M. et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352 (2019).
Yang, J. et al. Genomic inflation factors under polygenic inheritance. Eur. J. Human Genet. 19, 807–812 (2011).
Bulik-Sullivan, B. K. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
McLaren, W. et al. The Ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Gazal, S. et al. Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat. Genet. 49, 1421–1427 (2017).
The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Mancuso, N. et al. Probabilistic fine-mapping of transcriptome-wide association studies. Nat. Genet. 51, 675–682 (2019).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Do, T. P., Guo, S. & Ashina, M. Therapeutic novelties in migraine: new drugs, new hope? J. Headache Pain 20, 37 (2019).
Kurth, T. et al. Migraine and risk of cardiovascular disease in women: prospective cohort study. BMJ 353, i2610 (2016).
Hippisley-Cox, J., Coupland, C. & Brindle, P. Development and validation of QRISK3 risk prediction algorithms to estimate future risk of cardiovascular disease: prospective cohort study. BMJ 357, j2099 (2017).
Adelborg, K. et al. Migraine and risk of cardiovascular diseases: Danish population based matched cohort study. BMJ 360, k96 (2018).
Siewert, K. M. et al. Cross-trait analyses with migraine reveal widespread pleiotropy and suggest a vascular component to migraine headache. Int. J. Epidemiol. 49, 1022–1031 (2020).
Guo, Y. et al. A genome-wide cross-phenotype meta-analysis of the association of blood pressure with migraine. Nat. Commun. 11, 3368 (2020).
Nelson, C. P. et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 49, 1385–1391 (2017).
Loh, P.-R., Kichaev, G., Gazal, S., Schoech, A. P. & Price, A. L. Mixed-model association for biobank-scale datasets. Nat. Genet. 50, 906–908 (2018).
Pers, T. H. et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 6, 5890 (2015).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Need, A. C. & Goldstein, D. B. Schizophrenia genetics comes of age. Neuron 83, 760–763 (2014).
Gormley, P. et al. Common variant burden contributes to the familial aggregation of migraine in 1,589 families. Neuron 98, 743–753.e4 (2018).
Chasman, D. I. et al. Selectivity in genetic association with sub-classified migraine in women. PLoS Genet. 10, e1004366 (2014).
Nyholt, D. R. et al. Concordance of genetic risk across migraine subgroups: Impact on current and future genetic association studies. Cephalalgia 35, 489–499 (2014).
Ophoff, R. A. et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87, 543–552 (1996).
de Vries, B., Frants, R. R., Ferrari, M. D. & van den Maagdenberg, A. M. J. M. Molecular genetics of migraine. Human Genet. 126, 115 (2009).
Zhao, H. et al. Gene-based pleiotropy across migraine with aura and migraine without aura patient groups. Cephalalgia 36, 648–657 (2016).
Jacobs, B. & Dussor, G. Neurovascular contributions to migraine: moving beyond vasodilation. Neuroscience 338, 130–144 (2016).
Hoffmann, J., Baca, S. M. & Akerman, S. Neurovascular mechanisms of migraine and cluster headache. J. Cereb. Blood Flow Metab. 39, 573–594 (2017).
Bigal, M. E., Kurth, T., Hu, H., Santanello, N. & Lipton, R. B. Migraine and cardiovascular disease: possible mechanisms of interaction. Neurology 72, 1864–1871 (2009).
Malik, R. et al. Shared genetic basis for migraine and ischemic stroke: a genome-wide analysis of common variants. Neurology 84, 2132–2145 (2015).
Winsvold, B. S. et al. Genetic analysis for a shared biological basis between migraine and coronary artery disease. Neurol. Genet. 1, e10 (2015).
Mahmoud, A. N. et al. Migraine and the risk of cardiovascular and cerebrovascular events: a meta-analysis of 16 cohort studies including 1 152 407 subjects. BMJ open 8, e020498 (2018).
Daghlas, I., Guo, Y. & Chasman, D. I. Effect of genetic liability to migraine on coronary artery disease and atrial fibrillation: a Mendelian randomization study. Eur. J. Neurol. 27, 550–556 (2020).
Charles, A. The pathophysiology of migraine: implications for clinical management. Lancet Neurol. 17, 174–182 (2018).
Burstein, R., Noseda, R. & Borsook, D. Migraine: multiple processes, complex pathophysiology. J. Neurosci. 35, 6619–6629 (2015).
Andreou, A. P. & Edvinsson, L. Mechanisms of migraine as a chronic evolutive condition. J. Headache Pain 20, 117 (2019).
Olesen, J., Burstein, R., Ashina, M. & Tfelt-Hansen, P. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol. 8, 679–690 (2009).
Brandes, J. L. The influence of estrogen on migraine: a systematic review. JAMA 295, 1824–1830 (2006).
Borsook, D. et al. Sex and the migraine brain. Neurobiol. Dis. 68, 200–214 (2014).
Delaruelle, Z. et al. Male and female sex hormones in primary headaches. J. Headache Pain 19, 117 (2018).
Diener, H.-C. CGRP antibodies for migraine prevention — new kids on the block. Nat. Rev. Neurol. 15, 129–130 (2019).
de Vries, T., Villalón, C. M. & MaassenVanDenBrink, A. Pharmacological treatment of migraine: CGRP and 5-HT beyond the triptans. Pharmacol. Ther. 211, 107528 (2020).
Nelson, M. R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015).
King, E. A., Davis, J. W. & Degner, J. F. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 15, e1008489 (2019).
Benner, C. et al. Prospects of fine-mapping trait-associated genomic regions by using summary statistics from genome-wide association studies. Am. J. Human Genet. 101, 539–551 (2017).
Wainberg, M. et al. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 51, 592–599 (2019).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, s13742–s13748 (2015).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Tukiainen, T. et al. Chromosome X-wide association study identifies loci for fasting insulin and height and evidence for incomplete dosage compensation. PLoS Genet. 10, e1004127 (2014).
Mägi, R. & Morris, A. P. GWAMA: software for genome-wide association meta-analysis. BMC Bioinf. 11, 288 (2010).
Benner, C. et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501 (2016).
Westra, H.-J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
Zhernakova, D. V. et al. Identification of context-dependent expression quantitative trait loci in whole blood. Nat. Genet. 49, 139–145 (2017).
Ramasamy, A. et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 17, 1418–1428 (2014).
Grundberg, E. et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat. Genet. 44, 1084–1089 (2012).
Ng, B. et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat. Neurosci. 20, 1418–1426 (2017).
Fromer, M. et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 19, 1442–1453 (2016).
Võsa, U. et al. Large-scale cis and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
Kerimov, N. et al. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet. 53, 1290–1299 (2021).
Schmiedel, B. J. et al. Impact of genetic polymorphisms on human immune cell gene expression. Cell 175, 1701–1715.e16 (2018).
van der Wijst, M. G. P. et al. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet. 50, 493–497 (2018).
Wang, D. et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 362, eaat8464 (2018).
Price, A. L. et al. Long-range LD can confound genome scans in admixed populations. Am. J. Human Genet. 83, 132–139 (2008).
Lee, S. H., Wray, N. R., Goddard, M. E. & Visscher, P. M. Estimating missing heritability for disease from genome-wide association studies. Am. J. Human Genet. 88, 294–305 (2011).
Hujoel, M. L. A., Gazal, S., Hormozdiari, F., van de Geijn, B. & Price, A. L. Disease heritability enrichment of regulatory elements is concentrated in elements with ancient sequence age and conserved function across species. Am. J. Human Genet. 104, 611–624 (2019).
Trochet, H. et al. Bayesian meta-analysis across genome-wide association studies of diverse phenotypes. Genet. Epidemiol. 43, 532–547 (2019).
Cichonska, A. et al. metaCCA: summary statistics-based multivariate meta-analysis of genome-wide association studies using canonical correlation analysis. Bioinformatics 32, 1981–1989 (2016).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Hinrichs, A. S. et al. The UCSC genome browser database: update 2006. Nucleic Acids Res. 34, D590–D598 (2006).
Acknowledgements
We thank the study participants for their contribution to this research. We also thank the numerous individuals who contributed to sample collection, storage, handling, phenotyping and genotyping for each of the individual cohorts. We acknowledge the participants and investigators of the FinnGen study. This research has been conducted using the UK Biobank Resource under Application Number 22627. We are supported by following grants: the US National Institute of Neurological Disorders and Stroke (NINDS) of the US National Institutes of Health (NIH) (grant numbers R21NS09296 and R21NS104398 (D.I.C.)), the Finnish innovation fund Sitra and Finska Läkaresällskapet (E.W.), the Academy of Finland (grant nos. 288509, 312076, 336825 (M.P.)), the Sigrid Juselius Foundation (M.P. and S.R.), the Academy of Finland Center of Excellence in Complex Disease Genetics (grant no. 312062 (S.R.)), the Finnish Foundation for Cardiovascular Research (S.R.), University of Helsinki HiLIFE Fellow and Grand Challenge grants (S.R.), The Novo Nordisk Foundation (NNF14CC0001 and NNF17OC0027594 (T.F.H. and K.B.)), CANDY foundation (CEHEAD) (T.F.H.), and the South-Eastern Norway Regional Health Authority (grant no. 2020034 (B.S.W.)). A list of study-specific acknowledgements and funding information can be found in the Supplementary Note.
Author information
Authors and Affiliations
Consortia
Contributions
H.S., A.M.J.M.v.d.M., T.F.H. and M.P. conceived the study. H.H., B.S.W., S.E.R., G.B., A.V.E.H., L.J.A.K., L.F.T., R.N. and L.S.V. performed analyses in their respective cohorts. K. Hveem, H.S., K.S., A.M.J.M.v.d.M., T.F.H., S.R., J.-A.Z. and M.P. supervised analyses in their respective cohorts. H.H., B.S.W., D.I.C., D.R.N., A.M.J.M.v.d.M., T.F.H., J.-A.Z., A.P. and M.P. contributed to writing the manuscript. H.H. performed meta-analysis, and created figures and tables. H.H. and M.P. performed downstream analyses and drafted the manuscript. M.P. supervised project. All authors interpreted the results and reviewed and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
G.B., T.E.T., S.H.M., H.S. and K.S. are employees of deCODE genetics/Amgen. P.G. is a current employee and stockholder of GlaxoSmithKline but work was conducted while employed by Massachusetts General Hospital, Boston, MA, USA. T.F. reports possible competing interest for Electrocore (participation in clinical studies), Novartis (speakers’ honoraria, participation in advisory boards, participation in clinical studies), Teva (speakers’ honoraria, participation in advisory boards), Lilly (speakers’ honoraria, participation in clinical studies), and Bayer (speakers’ honoraria). M. Kallela has served on Advisory Boards for MSD and Allergan; has received funding for travel and/or speaker honoraria from MSD, Allergan, Teva, Novartis and Genzyme; has received compensation for producing educational material from Teva and Allergan; has received research support from Helsinki University Central Hospital; and holds stock/stock options and/or has received Board of Directors compensation from Helsinki Headache Center. T.K. reports having received honoraria from Eli Lilly, Newsenselab and Total for providing methodological advice and from the BMJ for editorial services. A.P. is the Scientific Director of the public–private partnership project FinnGen that has 12 industry partners that provide funding for the FinnGen project. V.A. has served on advisory board for Allergan, Lundbeck, Teva and Lilly. G.M.T. reports consultancy support from Novartis, Allergan, Lilly and Teva, and independent support from Dutch Organization for Scientific Research, the Dutch Heart & Brain Foundations, IRRF and Dioraphte. Other authors report no competing interests.
Peer review
Peer review information
Nature Genetics thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Note and Figs. 1–8.
Supplementary Table 1
Supplementary Tables 1–20.
Supplementary Data 1
Regional LocusZoom plots of the 123 independent migraine risk loci identified from the meta-analysis.
Supplementary Data 2
Forest plots of the 123 lead migraine variants.
Supplementary Data 3
Forest plots of 10 variants that have been previously reported to associate with migraine but failed to replicate in our study.
Supplementary Data 4
Forest plots of the 123 lead migraine variants from the MO meta-analysis.
Supplementary Data 5
Forest plots of the 123 lead migraine variants from the MA meta-analysis.
Supplementary Data 6
Subtype-specific combined log-odds-ratio estimates and posterior probabilities from subtype-specificity analysis for the 123 lead migraine variants.
Supplementary Data 7
Pairwise EAF and MAF plots against the reference cohort (UKBB).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hautakangas, H., Winsvold, B.S., Ruotsalainen, S.E. et al. Genome-wide analysis of 102,084 migraine cases identifies 123 risk loci and subtype-specific risk alleles. Nat Genet 54, 152–160 (2022). https://doi.org/10.1038/s41588-021-00990-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-021-00990-0
This article is cited by
-
Migraine - a borderland disease to epilepsy: near it but not of it
The Journal of Headache and Pain (2024)
-
Causal influences of neuropsychiatric disorders on Alzheimer’s disease
Translational Psychiatry (2024)
-
Oxidative Stress and Migraine
Molecular Neurobiology (2024)
-
Influences of Genetic and Environmental Factors on Chronic Migraine: A Narrative Review
Current Pain and Headache Reports (2024)
-
Alterations in DNA methylation associate with reduced migraine and headache days after medication withdrawal treatment in chronic migraine patients: a longitudinal study
Clinical Epigenetics (2023)
