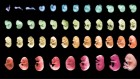- TECHNOLOGY FEATURE
- Correction 21 July 2021
- Correction 24 August 2021
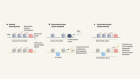
Single-cell analysis enters the multiomics age
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 595, 614-616 (2021)
doi: https://doi.org/10.1038/d41586-021-01994-w
Updates & Corrections
-
Correction 21 July 2021: An earlier version of the caption for the first image erroneously stated that the top section of the image represented single-cell data.
-
Correction 24 August 2021: An earlier version of this article erroneously stated that Le Cao’s students used a machine-learning approach called partial least squares. In fact, the team comprised one student and one postdoc, and the technique they used was an extension of the partial least squares method.
References
Argelaguet, R. et al. Nature 576, 487–491 (2019).
Cao, J. et al. Science 370, eaba7721 (2020).
Ma, S. et al. Cell 183, 1103–1116 (2020).
Qiu, C. et al. Preprint at bioRxiv https://doi.org/10.1101/2021.06.08.447626 (2021).
Argelaguet, R., Cuomo, A. S. E., Stegle, O. & Marioni, J. C. Nature Biotechnol. https://doi.org/10.1038/s41587-021-00895-7 (2021).
Bocchi, V. D. et al. Science 372, eabf5759 (2021).
Mimitou, E. P. et al. Nature Biotechnol. https://doi.org/10.1038/s41587-021-00927-2 (2021).
 Starfish enterprise: finding RNA patterns in single cells
Starfish enterprise: finding RNA patterns in single cells
 The secret life of cells
The secret life of cells
 Single-cell sequencing made simple
Single-cell sequencing made simple
 NatureTech hub
NatureTech hub