- NEWS AND VIEWS
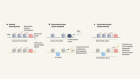
Cancer aided by greasy traitors
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 591, 204-206 (2021)
doi: https://doi.org/10.1038/d41586-021-00421-4
References
Sojka, D. K., Huang, Y.-H. & Fowell, D. J. Immunology 124, 13–22 (2008).
Lim, S. A. et al. Nature 591, 306–311 (2021).
Doi, T. et al. Clin. Cancer Res. 25, 6614–6622 (2019).
Pauken, K. E., Dougan, M., Rose, N. R., Lichtman, A. H. & Sharpe, A. H. Trends Immunol. 40, 511–523 (2019).
Goldstein, J. L., DeBose-Boyd, R. A. & Brown, M. S. Cell 124, 35–46 (2006).
Corn, K. C., Windham, M. A. & Rafat, M. Prog. Lipid Res. 80, 101055 (2020).
Overacre-Delgoffe, A. E. et al. Cell 169, 1130–1141 (2017).
Sanfilippo, K. M. et al. J. Clin. Oncol. 34, 4008–4014 (2016).
Alexandre, L. et al. Gastroenterology 150, 854–865 (2016).
Wu, B. U. et al. Am. J. Gastroenterol. 110, 1233–1239 (2015).
Juarez, D. & Fruman, D. A. Trends Cancer https://doi.org/10.1016/j.trecan.2020.11.008 (2021).
Berod, L. et al. Nature Med. 20, 1327–1333 (2014).
Favier, L. A. & Schulert, G. S. Appl. Clin. Genet. 9, 101–110 (2016).
Competing Interests
U.H.B. anticipates employment at Janssen later this year.
 Read the paper: Lipid signalling enforces functional specialization of Treg cells in tumours
Read the paper: Lipid signalling enforces functional specialization of Treg cells in tumours
 Teamwork by different T-cell types boosts tumour destruction by immunotherapy
Teamwork by different T-cell types boosts tumour destruction by immunotherapy
 A dendritic cell multitasks to tackle cancer
A dendritic cell multitasks to tackle cancer