- NEWS AND VIEWS
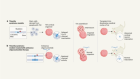
Mitochondrial genome editing gets precise
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 583, 521-522 (2020)
doi: https://doi.org/10.1038/d41586-020-01974-6
References
Russell, O. M., Gorman, G. S., Lightowlers, R. N. & Turnbull, D. M. Cell 181, 168–188 (2020).
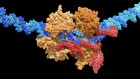
Mok, B. Y. et al. Nature 583, 631–637 (2020).
Salter, J. D. & Smith, H. C. Trends Biochem. Sci. 43, 606–622 (2018).
Moretton, A. et al. PLoS ONE 12, e0176795 (2017).
Schmidt, O., Pfanner, N. & Meisinger, C. Nature Rev. Mol. Cell Biol. 11, 655–667 (2010).
Gammage, P. A., Moraes, C. T. & Minczuk, M. Trends Genet. 34, 101–110 (2018).
Kunz, C., Saito, Y. & Schär, P. Cell. Mol. Life Sci. 66, 1021–1038 (2009).
Hellebrekers, D. M. E. I. et al. Hum. Reprod. Update 18, 341–349 (2012).
Bacman, S. R. et al. Nature Med. 24, 1696–1700 (2018).
Gammage, P. A. et al. Nature Med. 24, 1691–1695 (2018).
Greenfield, A. et al. Nature Biotechnol. 35, 1059–1068 (2017).
St. John, J. C., Facucho-Oliveira, J., Jiang, Y., Kelly, R. & Salah, R. Hum. Reprod. Update 16, 488–509 (2010).
 Read the paper: A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing
Read the paper: A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing
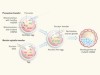 Replacing the cell’s power plants
Replacing the cell’s power plants
 Mitochondrial DNA can be inherited from fathers, not just mothers
Mitochondrial DNA can be inherited from fathers, not just mothers