
The coronavirus known as severe acute respiratory syndrome coronavirus 2 (SARS‑CoV-2) emerged in late 2019, and certain aspects of the disease it causes — COVID‑19 — continue to baffle clinicians and researchers. It is estimated that SARS-CoV-2 has already infected more than 9 million people and claimed more than 450,000 lives worldwide, and this pandemic has paralysed economies globally. Writing in Nature, Zhang et al.1 present data on the evolution of two major lineages of SARS-CoV-2, together with information regarding human-host determinants of disease severity from their analysis of 326 people in Shanghai, China, who were infected with SARS-CoV-2.
Read the paper: Viral and host factors related to the clinical outcome of COVID-19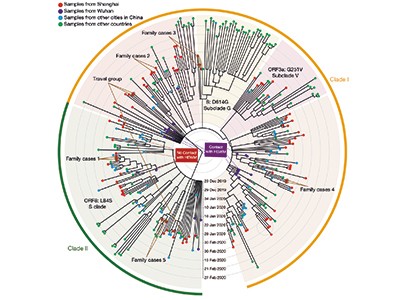
SARS-CoV-2, which caught the world by surprise, was initially thought to have ‘jumped’ to humans from an animal host at the Huanan Seafood Wholesale Market in Wuhan, China. When the first cases of a previously unknown disease, initially described as ‘a severe pneumonia with unknown aetiology’, were identified in Wuhan at the end of December 2019, the majority of cases could be traced back to this market. The implication was that the new coronavirus had crossed the species barrier at the market from an infected live animal on sale. The Malayan pangolin, a scaly anteater previously living in relative obscurity, suddenly faced allegations that it was the culprit, although whether this protected creature was on sale in the market at that time is uncertain (see Nature http://doi.org/ggpxhb; 2020). However, some cases of the disease in early December 2019 in Wuhan had no obvious links to the market2.
Zhang et al. analysed 94 complete genome sequences of SARS-CoV-2 in samples obtained from people living in Shanghai who had visited a health-care clinic in January or February 2020, and compared these data with 221 other sequences of the virus. The authors’ results reinforce previous observations3 of two major phylogenetic lineages (clades) of SARS-CoV-2 during the early phase of the outbreak in China. They are distinguished by two distinctive nucleotide differences, suggesting multiple origins for the human infections transmitted to people in Shanghai (which is about 800 kilometres by road from Wuhan).
The two lineages are termed clades I and II (Fig. 1). They presumably evolved independently from a common ancestor, but their ancestry in terms of how they relate to each other is unclear, because they differ at only two genomic sites. One difference involves a particular nucleotide in the sequence that encodes amino-acid residue number 84 in the viral protein ORF8. If the nucleotide has a thymine base (clade I), the sequence encodes the amino acid leucine; if it has a cytosine base (clade II), the sequence encodes a serine. The other difference is at a nucleotide in the gene ORF1ab, which contains either cytosine (clade I) or thymine (clade II); both the resulting nucleotide sequences encode serine.

Figure 1 | Assessing the relationship between coronavirus lineages and COVID-19 severity. Zhang et al.1 studied people from Shanghai, China, who were infected with the coronavirus SARS-CoV-2 in early 2020. a, Consistent with previous research3, the coronavirus genome sequences Zhang et al. identified belonged to two lineages, termed clade I and clade II. These differ at two nucleotides and probably evolved independently from a common ancestor. Clade I was associated with some cases linked to Huanan Seafood Wholesale Market in Wuhan, China, originally thought to have been the source of the outbreak, whereas the authors found clade II infections that did not have links to the market. Both lineages might have spread independently at the same time. b, Zhang and colleagues categorized the individuals into four groups, depending on their disease severity, which ranged from those unaffected by symptoms (the asymptomatic group) to the critical group (those requiring artificial ventilation to breathe). Both clades had the same ability to cause the different disease groupings. An increase in disease severity was accompanied by a depletion of immune cells called CD3+ T cells and an increase in the pro-inflammatory cytokine proteins IL-6 and IL-8. High cytokine levels can cause an intense immune response known as a cytokine storm.
Combining viral genomics with epidemiological evidence of how people might have picked up the infection, Zhang et al. show that the viral genomes from six people with established links to the Wuhan market cluster in clade I on the SARS-CoV-2 family tree, whereas the viral genomes of three cases without known links to the market cluster in clade II. These data support the idea that the market might not have been the origin of the pandemic. Instead, they suggest that clades I and II originated from a common viral ancestor and spread independently at the same time: clade I through the market and clade II outside it. Therefore, the animal-to-human transfer might have occurred elsewhere, seeding transmission chains that found their way to the market — where the high density of stalls and susceptible humans facilitated uncontainable spread in, and subsequently beyond, the site.
The circulation of different ‘types’ of SARS‑CoV-2 has been a contentious topic, stemming from the observation of distinct phylogenetic lineages. However, such genetic divergence among viruses, especially in the context of ‘immunologically naive’ human hosts (those who have never encountered the virus before) is expected. This can be explained by the ‘founder effect’, which is common during viral outbreaks — if a limited number of viral variants randomly enter a new geographical region where there is a susceptible population, their subsequent spread there facilitates the dominance of those variants at that location.
Going back in time for an antibody to fight COVID-19
However, the difference in prevalence of those variants in that particular population, compared with infected populations in other regions, does not necessarily equate to improved fitness of those variants in terms of viral replication and transmission4. Consistent with this idea, Zhang et al. find no evidence of any association between either of the two clades, or between any mutations in subclades, and the clinical parameters they assessed to categorize COVID-19 disease severity. Although this finding is not surprising, given that the two clades differ by only two nucleotides out of the approximately 30,000 nucleotides in the SARS-CoV-2 genome, it highlights the fact that distinct phylogenetic lineages do not necessarily indicate distinct viral strains with different disease outcomes.
Having found no difference in clinical outcomes between infections with the two SARS-CoV-2 lineages, Zhang et al. analysed various parameters of immune-system function in the human hosts to identity factors that contribute to disease severity.
The authors focused on four disease categories with well described definitions of clinical outcomes. The least-affected individuals were asymptomatic and had no fever, no breathing problems and no signs of lung damage on X-ray scans. Mild cases were those in people who had fever and signs of inflammation on X-rays of their lungs, indicating pneumonia. People with severe disease had difficulty breathing and had hallmarks of lung damage described as ‘ground-glass opacities’ on X-rays. Critically ill patients had acute respiratory distress syndrome, and required mechanical ventilation to assist breathing. In agreement with previous research5, Zhang and colleagues found that being older, the presence of other pre-existing medical conditions (termed comorbidities), and male gender were the leading factors associated with a higher probability of more-severe disease.
From the analysis of blood samples, the authors provide evidence of changes that characterized the severe and critical cases of COVID-19. One characteristic of these cases was lymphocytopenia — an abnormally low number of lymphocytes (a type of white blood cell involved in immune responses) in the blood. Zhang et al. attributed this lymphocytopenia to the depletion of a particular type of lymphocyte called a CD3+ T cell, most probably reflecting movement of these T cells from the blood to sites of infection in tissues.
CRISPR tool scales up to interrogate a huge line-up of viral suspects
Another characteristic of the severe and critical cases was abnormally high levels of the cytokines IL-6 and IL-8, which are small proteins that promote inflammation. High levels of pro-inflammatory cytokines drive an intense immune response that is commonly referred to as a cytokine storm. Immune-system cells called macrophages, which are present in the lung, can make IL-6 and IL-8, and are often the initial cellular mediators of a cytokine storm in other respiratory infections. However, the precise cell populations contributing to the prolonged cytokine storm that occurs in some cases of COVID-19 remain to be defined.
The inverse correlation between high levels of IL-6 or IL-8 and low lymphocyte numbers hints at underlying mechanisms that might link these characteristics of severe disease. The possibility that high cytokine levels cause lymphocytopenia is consistent with the observation that people with COVID-19 who were treated with the drug tocilizumab, which blocks IL-6-mediated signalling, had their bloodstream levels of lymphocytes restored to nearer normal6. However, further experimental and mechanistic studies are needed to establish whether a causal connection underlies the correlation between these cytokine levels and lymphocytopenia. Of note is the discordant time frame of changes in these two parameters — T-cell depletion is evident from the first week of overt disease, whereas a cytokine storm arises later, when COVID-19 has become severe.
Moreover, neither lymphocytopenia nor a cytokine storm are exclusive to COVID-19. Both are hallmarks of many types of severe respiratory infection, including human infection by avian influenza viruses, and severe acute respiratory syndrome (SARS), a disease caused by a coronavirus related to SARS-CoV-2. To delineate the immunological signatures that are specific to COVID-19, more-detailed cellular and molecular analyses will be needed.
Tracing the evolution of SARS-CoV-2 is fundamental for informing the public-health policies needed to limit disease spread. Dissecting the underlying causes and mechanisms of perturbed immune defences, such as the depletion of CD3+ T cells and the heightened pro-inflammatory response, as well as determining the crucial clinical and molecular hallmarks of COVID-19, are of paramount importance for the design of treatment strategies and effective vaccines. Zhang et al. lay some essential groundwork that should aid in these Herculean tasks, and their work raises key questions that will need to be answered if we are to limit this pandemic and try to prevent a future one.
 Read the paper: Viral and host factors related to the clinical outcome of COVID-19
Read the paper: Viral and host factors related to the clinical outcome of COVID-19
 Going back in time for an antibody to fight COVID-19
Going back in time for an antibody to fight COVID-19
 CRISPR tool scales up to interrogate a huge line-up of viral suspects
CRISPR tool scales up to interrogate a huge line-up of viral suspects





