- NEWS AND VIEWS
Tension in tumour cells keeps metabolism high
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 578, 517-518 (2020)
doi: https://doi.org/10.1038/d41586-020-00314-y
References
Butcher, D. T., Alliston, T. & Weaver, V. M. Nature Rev. Cancer 9, 108–122 (2009).
Zanotelli, M. R. et al. Mol. Biol. Cell 29, 1–9 (2018).
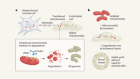
Park, J. S. et al. Nature 578, 621–626 (2020).
Vander Heiden, M. G. & DeBerardinis, R. J. Cell 168, 657–669 (2017).
Sullivan, W. J. et al. Cell 175, 117–132 (2018).
Bertero, T. et al. Cell Metab. 29, 124–140 (2019).
Seo, B. R. et al. Sci. Transl. Med. 7, 301ra130 (2015).
Hu, H. et al. Cell 164, 433–446 (2016).
Bays, J. L., Campbell, H. K., Heidema, C., Sebbagh, M. & DeMali, K. A. Nature Cell Biol. 19, 724–731 (2017).
Pickup, M. W., Mouw, J. K. & Weaver, V. M. EMBO Rep. 15, 1243–1253 (2014).
Burgstaller, G. et al. Eur. Respir. J. 50, 1601805 (2017).
Kai, F., Drain, A. P. & Weaver, V. M. Dev. Cell 49, 332–346 (2019).
Samuel, M. S. et al. Cancer Cell 19, 776–791 (2011).
Paszek, M. J. et al. Cancer Cell 8, 241–254 (2005).
Lipkowitz, S. & Weissman, A. M. Nature Rev. Cancer 11, 629–643 (2011).
Laklai, H. et al. Nature Med. 22, 497–505 (2016).
 Read the paper: Mechanical regulation of glycolysis via cytoskeleton architecture
Read the paper: Mechanical regulation of glycolysis via cytoskeleton architecture
 Metabolic signal curbs cancer-cell migration
Metabolic signal curbs cancer-cell migration
 Metabolic vulnerability in tumours illuminated
Metabolic vulnerability in tumours illuminated