
- NEWS AND VIEWS
Malaria parasites fine-tune mutations to resist drugs
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 576, 217-219 (2019)
doi: https://doi.org/10.1038/d41586-019-03587-0
References
World Health Organization. World Malaria Report 2018 (2018).
Haldar, K., Bhattacharjee, S. & Safeukui, I. Nature Rev. Microbiol. 16, 156–170 (2018).
Kim, L. et al. Nature 576, 315–320 (2019).
Goldberg, D. E. Curr. Top. Microbiol. Immunol. 295, 275–291 (2005).
van der Pluijm, R. W. et al. Lancet Infect. Dis. 19, 952–961 (2019).
Ross, L. S. et al. Nature Commun. 9, 3314 (2018).
Fidock, D. A. et al. Mol. Cell 6, 861–871 (2000).
Summers, R. L. et al. Proc. Natl Acad. Sci. USA 111, E1759–1767 (2014).
Martin, R. E. & Kirk, K. Mol. Biol. Evol. 21, 1938–1949 (2004).
Nogales, E. & Scheres, S. H. Mol. Cell 58, 677–689 (2015).
Pelleau, S. et al. Proc. Natl Acad. Sci. USA 112, 11672–11677 (2015).
Conrad, M. D. & Rosenthal, P. J. Lancet Infect. Dis. 19, e338–e351 (2019).
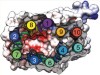 Read the paper: Structure and drug resistance of the Plasmodium falciparum transporter PfCRT
Read the paper: Structure and drug resistance of the Plasmodium falciparum transporter PfCRT
 Spotlight on proteins that aid malaria
Spotlight on proteins that aid malaria
 Battling disease by giving mosquitoes an antimalarial drug
Battling disease by giving mosquitoes an antimalarial drug


