- NEWS AND VIEWS
A powerful cell-protection system prevents cell death by ferroptosis
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 575, 597-598 (2019)
doi: https://doi.org/10.1038/d41586-019-03145-8
References
Crane, F. L. Mitochondrion 7 (Suppl.), S2–S7 (2007).
Doll, S. et al. Nature 575, 693–698 (2019).
Bersuker, K. et al. Nature https://doi.org/10.1038/s41586-019-1705-2 (2019).
Dixon, S. J. et al. Cell 149, 1060–1072 (2012).
Stockwell, B. R. & Jiang, X. Cell Metab. 30, 14–15 (2019).
Hirschhorn, T. & Stockwell, B. R. Free Radic. Biol. Med. 133, 130–143 (2019).
Stockwell, B. R. et al. Cell 171, 273–285 (2017).
Yang, W. S. et al. Cell 156, 317–331 (2014).
Liu, H., Schreiber, S. L. & Stockwell, B. R. Biochemistry 57, 2059–2060 (2018).
Shimada, K. et al. Nature Chem. Biol. 12, 497–503 (2016).
Competing Interests
B.R.S. holds equity in and serves as a consultant to Inzen Therapeutics, is an inventor on patents and patent applications related to ferroptosis, and has consulted with additional pharmaceutical and biotechnology companies in the past.
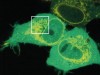 Read the paper: FSP1 is a glutathione-independent ferroptosis suppressor
Read the paper: FSP1 is a glutathione-independent ferroptosis suppressor
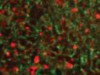 Read the paper: The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis
Read the paper: The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis
 Survival skills ensure that cancer spreads
Survival skills ensure that cancer spreads
 Cancer-cell death ironed out
Cancer-cell death ironed out