- NEWS AND VIEWS
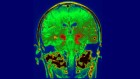
Brain-to-pancreas signalling axis links nicotine and diabetes
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 574, 336-337 (2019)
doi: https://doi.org/10.1038/d41586-019-02975-w
References
Chiolero, A., Faeh, D., Paccaud, F. & Cornuz, J. Am. J. Clin. Nutr. 87, 801–809 (2008).
Chase, H. P. et al. J. Am. Med. Assoc. 265, 614–617 (1991).
Madsbad, S. et al. Diabetes Care 3, 41–43 (1980).
Will, J. C., Galuska, D. A., Ford, E. S., Mokdad, A. & Calle, E. E. Int. J. Epidemiol. 30, 540–546 (2001).
Benowitz, N. L. N. Engl. J. Med. 362, 2295–2303 (2010).
Duncan, A. et al. Nature 574, 372–377 (2019).
Liu, Z. & Habener, J. F. J. Biol. Chem. 283, 8723–8735 (2008).
Picciotto, M. R., Addy, N. A., Mineur, Y. S. & Brunzell, D. H. Prog. Neurobiol. 84, 329–342 (2008).
Khiroug, L., Sokolova, E., Giniatullin, R., Afzalov, R. & Nistri, A. J. Neurosci. 18, 2458–2466 (1998).
Centers for Disease Control and Prevention. MMWR Morbid. Mortal. Wkly Rep. 60, 1513–1519 (2011).
Solberg, L. I., Desai, J. R., O’Connor, P. J., Bishop, D. B. & Devlin, H. M. Ann. Fam. Med. 2, 26–32 (2004).
Lee, S., Lee, C. E., Elias, C. F. & Elmquist, J. K. J. Comp. Neurol. 517, 925–939 (2009).
Tweed, J. O., Hsia, S. H., Lutfy, K. & Friedman, T. C. Trends Endocrinol. Metab. 23, 334–342 (2012).
Pirie, P. L., Murray, D. M. & Luepker, R. V. Am. J. Public Health 81, 324–327 (1991).
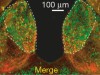 Read the paper: Habenular TCF7L2 links nicotine addiction to diabetes
Read the paper: Habenular TCF7L2 links nicotine addiction to diabetes
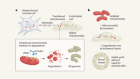
 Location matters for insulin-producing cells
Location matters for insulin-producing cells
 Weight loss through smoking
Weight loss through smoking